3
Insulin resistance pathogenesis in visceral fat and gut organisms
Epidemiological work has shown that visceral adiposity is strongly related to metabolic disorders including insulin resistance. What regulates visceral fat deposition and why it is so metabolically deleterious remains largely unclear. Recent data suggest that the gastrointestinal tract may be a central player in the development of visceral fat accumulation and metabolic syndrome. An impaired gut barrier function, as a consequence of inflammation and/or altered microbiota composition, increases the leak of microbial molecules and their metabolites to the adjacent mesenteric fat resulting in hypertrophy and inflammation of the fat depot. Subsequently, the increased efflux of fatty acids and pro-inflammatory factors in the portal vein leads to liver dysfunction and systemic insulin resistance.
Obesity is a condition in which fat accumulation in adipose tissue is in excess to an extent that health may be impaired. Obese individuals are at an increased risk of developing chronic health problems including cardiovascular disease, type 2 diabetes, hypertension, non-alcoholic fatty liver disease and certain cancers [1]. A subset of obese individuals, classified as ‘metabolically healthy obese’ (MHO) account for ~20% of the obese population, remain insulin-sensitive and appear to be less susceptible to obesity-related metabolic complications [2]. It has been estimated that type 2 diabetes and cardiovascular disease is six- and twofold respectively more common in ‘at-risk’ obese as compared to MHO individuals [3]. An important feature of MHO individuals is they have proportionally less visceral fat (the abdominal fat within the visceral cavity). This is consistent with recent data suggesting that regional fat distribution is an important determinant of insulin sensitivity and metabolic risk [4]. What regulates visceral fat deposition and why it is so metabolically dangerous remains largely unclear. This article summarises literature on underlying mechanisms of visceral adipose dysfunction and the emerging role of the gut, and its resident microbes, as a central player in metabolic disorders (Figure 1). 47
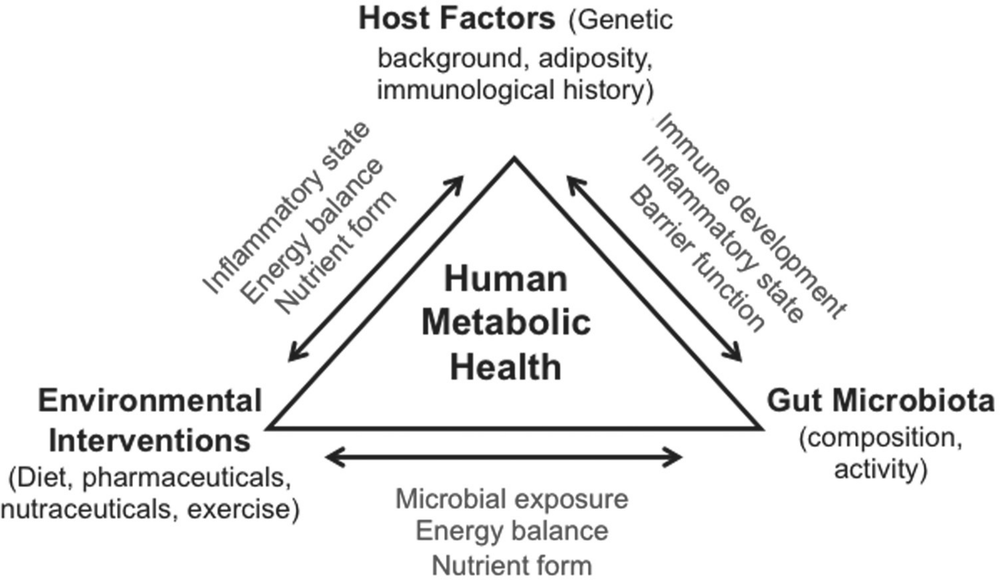
Figure 1. Overview of interconnections between lifestyle factors, host factors and gut microbiota in metabolic health. Refer to the text for a more detailed explanation and description of terms used.
Regional fat distribution
The distribution of adipose tissue varies considerably among individuals even with similar total body fat. What regulates regional fat deposition is not entirely clear but is at least known to be affected by gender, age and ethnicity. Men tend to have more visceral fat and have at least twice the proportion of fat localised in the intra-abdominal depot as compared to women [5, 6]. The gender-specific difference in fat distribution, however, appears to diminish in older age as females tend to develop central adiposity after menopause [7]. Ethnicity also affects regional adiposity. Aboriginal men and women in Australia have been shown to have greater waist-to-hip ratio as compared to their European Australian counterparts and the difference is observed across all BMI levels up to 30 kg/m2 [8]. Central obesity is also more common in Hispanic as compared to white women in early adulthood [9].
Epidemiological data suggest a relationship between central adiposity and metabolic risk factors including elevated blood pressure, fasting plasma glucose and triglycerides [10]. Visceral fat accounts for ~50% of the variance in insulin sensitivity [11, 12] and has been shown to be a predictor for future insulin resistance [13]. The accumulation of visceral fat is strongly related to reduced insulin responsiveness irrespective of adiposity [6, 14]. Conversely, the association between visceral fat reduction and improved insulin sensitivity has been consistently demonstrated in obese [15], glucose intolerance-impaired [16] and type 2 diabetic [17] individuals.48
In contrast to visceral fat, the relationship between subcutaneous fat and metabolic risk is less clear-cut. Wagenknechi and co-workers [18] reported that both visceral and subcutaneous adiposity were inversely associated with insulin sensitivity; Cnop et al. [19] estimated that subcutaneous fat only accounted for 5% of the variance in insulin sensitivity; in patients with type 2 diabetes, Miyazaki and colleagues [20] reported that insulin-stimulated glucose disposal was inversely correlated with visceral but not subcutaneous fat area; data from the Framingham Heart Study even suggested a protective effect of subcutaneous fat against metabolic and cardiovascular risk in individuals in the highest tertile of visceral adiposity [21]. Some attributed the inconsistent relationship between subcutaneous fat and insulin sensitivity to different metabolic effects of the subdivisions of the fat depot [22], with the deep subcutaneous adipose tissue exhibiting a secretory profile similar to that of visceral fat [22]. It has also been proposed that once the accumulation of visceral adipose tissue exceeds a certain threshold, the contribution of the depot to insulin resistance would overwhelm that of abdominal subcutaneous fat regardless of subdivisions [23].
It is logical to hypothesise that the intrinsic difference(s) between visceral and subcutaneous adipocytes (fat cells) may contribute to the region-specific metabolic effects of fat depots. Indeed, visceral adipocytes are shown to be both structurally and functionally distinct. For example, they are larger in size, less insulin-sensitive and have a greater lipolytic activity as compared to subcutaneous adipocytes (for details please refer to a comprehensive review by Ibrahim [24]). These characteristics, however, do not appear to completely account for the deleterious nature of visceral fat.
Obesity, inflammation and insulin resistance
Over the past decade it has been increasingly recognised that adipose tissue is a complex endocrine organ secreting, inter alia, a range of cytokines [25]. Together with the well-characterised state of low-grade chronic inflammation in obesity [26], this points to an entirely novel angle to investigate regional metabolic effects of fat depots. Adipose tissue produces a range of protein factors including cytokines, chemokines and growth factors. Leptin and adiponectin increase insulin sensitivity; tumor necrosis factor (TNF)-alpha, interleukin (IL)-1beta, IL-6, IL-8 and monocyte chemoattractant protein (MCP)-1 are pro-inflammatory, either by direct activation of the inflammatory signalling pathway or by promoting the migration of immune cells; IL-10, which inhibits the production of pro-inflammatory cytokines, is one of the main adipose-derived anti-inflammatory factors.
Epidemiological data indicate an association between chronic inflammation and decreased insulin sensitivity. Circulating levels of inflammatory markers are increased in individuals with type 2 diabetes, insulin resistance or the metabolic syndrome [27]. In a prospective case-control study, elevated plasma levels of IL-6 and C-reactive protein were shown to be associated with an increased risk of developing type 2 diabetes independent of BMI, physical activity and other lifestyle factors [28]. The role of inflammation in the pathogenesis of insulin resistance is further supported by the effect of high-dose aspirin, an anti-inflammatory drug commonly used to treat rheumatoid arthritis, in reducing 49fasting blood glucose and improving insulin-stimulated peripheral glucose uptake in type 2 diabetic patients [29].
More importantly, there is evidence supporting inflammation as the major determinant of the deleterious metabolic effects of visceral fat. Compared to people with normal fat distribution, the plasma concentrations of inflammatory mediators are up to ~50% higher in centrally obese individuals [30]. Direct comparison of cytokine production using adipose tissue explants in vitro revealed that visceral fat released higher concentrations of pro-inflammatory cytokines including IL-6, IL-8 and TNF-alpha as compared to subcutaneous fat [31, 32]. Using adipose tissue-conditioned media, we provided direct evidence for visceral fat induction of insulin resistance in skeletal muscle in vitro [33]. Individual pro-inflammatory cytokines, specifically IL-6 [33] IL-1beta [34] and TNF-alpha [35], have been shown to inhibit insulin signalling. Further, our data suggest that the sequential activation of nuclear factor kappa B (NFκB) and mammalian target of rapamycin complex 1 (mTORC1) may be the common pathway which mediates visceral fat-induced insulin resistance [33]. Briefly, pro-inflammatory cytokines phosphorylates inhibitor of kappa B kinase (IKK) and activates mTORC1. Ribosomal S6 kinase 1, a downstream effector of mTORC1, phosphorylates insulin receptor substrate-1 and inhibits its interaction with the insulin receptor and/or p85 subunit of phsophatidylinositol 3-kinase. Also, activated IKK degrades inhibitor protein inhibitor kappa B. The subsequent nuclear translocation of NFκB induces the transcription of pro-inflammatory cytokines and therefore provides a positive feedback to the inflammation cascade.
Macrophage infiltration in adipose tissue
Ameliorating fat inflammation has thus become a major focus in both prevention and treatment of type 2 diabetes. It is now recognised that in adipose the majority of cytokines originate from ‘non-fat’ cells [36]. Obesity is characterised by an increased accumulation of adipose tissue macrophages (ATMs) [37] which have been identified as the major contributor of both pro- and anti-inflammatory cytokines. Further, Harman-Boehm et al. [38] reported that the number of ATM was approximately two- to fourfold higher in omental (a major visceral fat depot in human) as compared to subcutaneous fat irrespective of levels of adiposity. A causal relationship between ATM infiltration and insulin resistance has been demonstrated in animal studies. Attenuating ATM infiltration, by genetic modification or pharmacological treatment, partially improved glucose homeostasis and insulin sensitivity in diet-induced obese mice, an effect associated with reduced expression of pro-inflammatory cytokines [39]. Conversely, over-expression of MCP-1, a major chemokine which promotes macrophage infiltration, in adipocytes increased ATM abundance and induced insulin resistance without affecting adipose tissue weight [40].
Mechanisms of macrophage infiltration
The mechanisms by which ATM infiltration occurs, however, are not entirely clear. It has long been proposed that macrophage infiltration is part of an immune response to adipose dysfunction in obesity. A credible hypothesis is that chronic energy excess and increased 50lipid accumulation leads to adipocyte hypertrophy. Limited lipid storage capacity then induces oxidative stress, which results in necrotic-like cell death and subsequently triggers an inflammatory response [41].
The increased susceptibility of visceral adipocytes to cell death may lead to differential ATM infiltration. The majority of ATMs aggregate around dead adipocytes and form the characteristic ‘crown-like structures’ (CLS). In genetically (ob/ob and db/db) and diet-induced obese mice, both dead adipocytes and CLS are more abundant in visceral as compared to subcutaneous fat [42, 43]. A linear correlation between adipocyte size and CLS density has been demonstrated in all fat depots, suggesting that visceral adipocytes may have a smaller critical size triggering death and therefore promotes the migration of macrophages into this fat depot [43].
Adipocyte hypertrophy: the role of extracellular matrix
It is tempting to hypothesise that ATM infiltration, and therefore the associated deleterious metabolic consequences, may be preventable if the fat depots could expand indefinitely. The ability of adipocytes to expand is partly restricted by the extracellular matrix (ECM) and the abundance of ECM proteins determines the physical limit to cell growth. An increased area of fibrosis has been shown in adipose tissue from obese individuals as compared to lean controls [44]. More importantly, the mRNA (Messenger RNA) expression of collagen VI alpha3-subunit, the predominant ECM component in adipose tissue, is positively correlated with visceral fat content but no such relationship exists with the subcutaneous depot [45]. Khan and colleagues [46] used a genetic model of collagen VI disruption and demonstrated that the weakening of ECM structure allowed ‘stress-free’ expansion of adipocytes during high-fat feeding, an effect associated with a reduction in ATM infiltration and an improvement in glucose tolerance. In support, correlation between collagen VI and macrophage expression in adipose tissue and their inverse relationship with insulin sensitivity has also been recently demonstrated in humans [46].
The fact that obesity is characterised by both an increase in adipocyte size and ECM protein abundance in the adipose tissue, however, is intriguing and appears to work against the above-mentioned hypothesis. It is possible that, in the case of obesity, the increase in ECM component is a secondary effect from the already hypertrophic adipocytes in an attempt to restrict further lipid accumulation in the tissue. Mere adipocyte expansion is not physiologically viable in the long term due to excessive demands on the endoplasmic reticulum (ER) for protein folding, lipid esterification and nutrient-sensing results in ER stress [47]. This triggers an inflammatory response including the activation of the mitogen-activated protein kinase (MAPK) signalling pathway which has been linked to insulin resistance [48]. Further, unresolved ER stress and elevated intracellular levels of free fatty acids generate oxidative stress in the mitochondria which further impairs cellular function of adipocytes and may eventually induce apoptotic and/or necrotic cell death [49].
ECM regulates adipocyte size, and there is some evidence that ‘healthy’ hypertrophy may ameliorate adipose inflammation and obesity-associated insulin resistance. Thus, 51modulating ECM may provide some benefits during early obesity – when intervention precedes the stage at which adipocyte expansion becomes dangerous. However, such an approach would only achieve maximal long-term benefits when treatments to prevent further energy surplus and adipose expansion are in place.
Phenotypic switching of macrophages
The metabolic effects of ATMs are specific to their phenotypes. Macrophages are broadly classified as M1 (classically activated) or M2 (alternatively activated) based on the expression of cell surface markers. M1 macrophages produce primarily pro-inflammatory cytokines, eg IL-1beta, IL-6 and TNF-alpha, whereas M2 macrophages (which may be further subdivided into M2a, M2b and M2c) are generally responsible for tissue remodelling and down-regulation of an inflammatory response [50]. Obese mice exhibited an increased M1:M2 ATM ratio in visceral as compared to subcutaneous fat [51] and similar findings have also been reported in humans [52], suggesting the predominant effect of M1 ATMs in the pro-inflammatory nature of visceral fat. Recent data, however, challenge the simple M1/M2 classification system. For instance, CD11c has long been recognised as a typical M1 marker. In a study by Li and colleagues [50] in which mice were switched from a high-fat to a normal chow diet, the abundance of CD11c+ ATMs remained unchanged despite a reduction in the release of pro-inflammatory cytokines. Similar alterations in gene expression profile of CD11c+ ATMs have also been observed during the course of high-fat feeding [53], suggesting that such macrophages may exhibit a spectrum of functionality. Accordingly, the increased ATM infiltration in visceral fat does not necessarily, by itself, result in a ‘pro-inflammatory’ fat depot.
What further complicates our understanding of ATMs is their plasticity. The secretory function of macrophages is dependent on the specific microenvironment. It has been shown that macrophages activation, as defined by the expression of both cell surface markers and chemokines, is plastic and fully reversible depending on the presence and withdrawal of specific stimuli [54]. As in the case of chronic systemic inflammation in viscerally obese individuals, this would implicate the presence of factors that activate and maintain the ATMs in the pro-inflammatory phenotype. The nature and range of these factors is currently only poorly understood.
The role of gut in metabolic dysfunction
From leaky gut to visceral adipose expansion
The role of the gut in adipose physiology has long been recognised in patients with Crohn’s disease (a condition characterised by severe gut inflammation). These patients have an increased ratio of intra-abdominal to total abdominal fat as compared to healthy individuals [55]. The excess accumulation of mesenteric fat around the inflamed gut, known as ‘fat wrapping’, is associated with the prognosis of the disease [56] and is characterised by the increased infiltration of immune cells (eg macrophages and T-cells) and production of pro-inflammatory factors (eg IL-6 and MCP-1) [57]. Data from experimental models suggest causality between gut inflammation and mesenteric fat dysfunction – rats with induced 52colitis have 35% more mesenteric fat as compared to controls [58]. Using a similar model, Thomaz and colleagues [59] showed that mesenteric fat in the colitis animals had increased expression of F4/80 (a macrophage marker) and TNF-alpha. Importantly, it has been shown that the effect of gut inflammation on adipose tissue is localised [58] and therefore implicates anatomical proximity as important for gut-visceral fat interactions.
Indeed, there is some evidence suggesting that gut-induced adipose dysfunction may be a consequence of the direct ‘leakage’ of luminal antigens, microbiota and their metabolites through the gut wall into the adjacent mesenteric fat. Gut barrier integrity is normally maintained by multiple mechanisms. First, tight-junction proteins (eg zona occludens [ZO]-1, occludins and claudins) form multi-protein complexes to seal the space between neighbouring epithelial cells and therefore act as a physical barrier [60]. Second, intestinal epithelial cells produce a wide range of anti-microbial peptides, including defensins and cathelicidins, which serve as an immunological barrier to protect the mucosal surface from microbial pathogens [61]. Inflammation impairs gut barrier function, as evident by the increased gut permeability in patients with Crohn’s disease [62] and animal models of induced inflammation [63]. In vitro, activation of the inflammatory NFκB pathway (eg by TNF-alpha [64] and IL-1beta [65]) has been shown to disrupt tight-junction integrity by increasing the expression and activity of myosin light chain kinase, leading to the contraction of peri-junctional actin-myosin filaments and opening of the tight-junctions. This may result in a ‘leak’ of bacteria and their products, as demonstrated by the translocation of bacteria into the mesenteric fat in mice with induced-gut inflammation [63].
Mesenteric fat, therefore, is left to cope with an increased microbial load from the ‘leaky’ gut. Lipopolysaccharides (LPS), a major bacterial cell component derived from the cell wall of Gram-negative bacteria, induce insulin resistance in adipocytes [66]. Further, LPS induces the release of MCP-1 and pro-inflammatory cytokines in adipocytes [67, 68] and promotes the ‘pro-inflammatory’ polarisation of macrophages (ie increase production of IL-1, IL-6 and TNF-alpha and reduce that of the anti-inflammatory IL-10 [69]) and, therefore results in an inflamed fat depot. The chronic stimulation from the bacterial antigens also leads to activation and the subsequent enlargement of lymph nodes [70, 71], which together with the direct effect of bacterial stimuli on activating peroxisome proliferator-activated receptor-γ and then on to adipogenesis [71], results in mesenteric hypertrophy and/or hyperplasia. Mesenteric fat expansion as a consequence of the microbial leak from the inflamed gut however, may be an important defensive mechanism to prevent further translocation of bacteria and/or their products into the visceral cavity, which in extreme cases, can be fatal.
From leaky gut to systemic dysfunction
Unfortunately the metabolic consequences of a leaky gut do not stop at visceral adipose dysfunction. Following on from the inflamed and hypertrophic mesenteric fat, more pro-inflammatory factors and free fatty acids (as a result of increased lipolysis of insulin-resistant adipocytes) enter the portal circulation and subsequently lead to an inflamed, steatotic and insulin-resistant liver [72]. The deleterious effects of a diseased liver on carbohydrate and lipid homeostasis are obvious – reduced glucose uptake, impaired suppression of postprandial glucose release and over-production of fatty acids [73].53
There is also the effect of bacterial components and metabolites on systemic host metabolism when they enter the circulation. Under normal circumstances, only a very small amount of endotoxins (primarily LPS) pass through the gut barrier and reach the liver in which they are detoxified [74]. An impaired gut barrier function, however, will see an increased delivery of LPS into the liver. This may saturate the hepatic detoxification capacity and result in an ‘overflow’ of LPS into the systemic circulation [75]. A study by Pastor Rojo et al. [76] showed that 48% of patients with Crohn’s disease had an increased serum concentration of endotoxins. Recently, there is also some evidence for an elevated circulating level of endotoxins in overweight/obese individuals [77] and patients with type 2 diabetes [78]. This phenomenon, often referred to as ‘metabolic endotoxemia’, is associated with insulin resistance, chronic systemic inflammation and increased cardiovascular risk [79]. The metabolic consequence of endotoxemia is directly demonstrated in a study by Mehta and co-workers [80], in which intravenous LPS administration in healthy humans resulted in elevated plasma concentrations of inflammatory markers and a 35% reduction in insulin sensitivity. The molecular pathways by which LPS induces inflammation are detailed in a comprehensive review by Lu and colleagues [81]. Briefly, LPS is first recognised in the circulation by the LPS-binding protein and is then transported to the target cells, where LPS binds to CD14 and the toll-like receptor (TLR)-4/MD-2 receptor complex. After interacting with a series of adaptor proteins including myeloid differentiation primary response gene (MyD)-88, the net response to LPS is activation of both the NFκB and the MAPK signalling pathways and subsequently induction of the expression of pro-inflammatory cytokines. It should be noted that TLR4 is ubiquitously expressed in insulin-targeting tissues, eg adipose tissue, liver, skeletal muscle and pancreatic beta-cells, and there is evidence for TLR4-induced inflammation to inhibit insulin signal transduction (for details please refer to a review by Kim and Sears [82]).
In summary, the initially ‘localised’ inflammation of the gut may have deleterious consequence on whole-body metabolism. Inflammation and the associated insulin resistance, eg in liver, adipose tissue and skeletal muscle, will then stimulate insulin secretion from the beta-cells and subsequently results in peripheral insulin resistance and a vicious cycle of systemic metabolic dysfunction. This inflammatory response involves interaction with the gut microbiota.
Gut microbiota
Available evidence indicates that gut microbiota influence metabolic health in a variety of ways. The gastrointestinal tract (GIT) harbours a large microbial community (total of ca 1014 cells) with very high microbial cell densities in the ileum and large intestine [83, 84]. Many of the processes that occur in the GIT are either encoded by microbial genomes, or strongly influenced by microbial activity, and our physiology is a convergent of human and microbial traits [85, 86]. Accordingly, we need to consider the role of gut microbial community in the pathogenesis of metabolic disorders. Each individual’s gut microbiota is unique. While 80%–90% of the gut bacteria belong to the phyla Firmicutes and Bacteroidetes, the species involved and their relative abundances vary from person to person [87]. This variation in 54microbiota composition is widely accepted to be a contributing factor to differences in host physiological outcomes.
Gut microbiota and barrier function
Functional disruptions to the epithelial lining of GIT is characterised by an altered microbial community. In fact, epithelial cells and resident bacteria are thought to be synergistic partners in modulating gut barrier function. Gut microbiota contributes to barrier function through three different mechanisms. Firstly, normal mucosal resident bacteria competitively exclude other, potentially pathogenic bacteria, from attachment to the epithelial mucosa. Secondly, some gut bacteria have been shown to promote tight-junction integrity by inducing the expression levels of tight-junction-related genes [88] and/or by promoting the localisation of proteins (eg ZO-1 and occludin) in the tight-junctions [89]. Thirdly, gut bacteria produce substrates for the maintenance of enterocytes. Butyrate, for example, is the primary energy source for colonic epithelial cells. In germ-free mouse models, the absence of microbial butyrate resulted in the depletion of ATP level and induced autophagy, an effect reversed by introducing exogenous butyrate or by colonising germ-free mice with butyrate-producing bacteria [90]. These data support the notion that gut microbes are directly involved in the normal functioning of epithelial cells and maintenance of gut barrier integrity.
It has long been postulated that the beneficial effect of gut bacteria is not universal but is confined to specific species with other species being detrimental. To date only very few species are thoroughly investigated. Initial studies focused on organisms commonly isolated from the gut epithelium, eg Lactobacillus and Bifidobacterium. In gnotobiotic studies, Lactobacillus acidophilus has been shown to inhibit cell association and the invasion of flagellated bacteria, therefore ameliorating inflammation and improving gut barrier function [91]. Similarly, Bifidobacterium infantis increased epithelial integrity and was protective against inflammation-induced impaired gut barrier function both in vitro and in an experimental model of spontaneous colitis [92]. The effect of the described species (and strains) on maintaining epithelial barrier integrity in simple models (eg monoassociated gnotobionts), however, does not necessarily translate to physiological benefits in the natural gut system with its complex community. Lactobacillus and Bifidobacterium only account for a small proportion of the gut microbial community and therefore their metabolic effects may be relatively minor as compared to that of the more abundant genera such as Clostridium.
Recent studies have focused on investigating the function of gut microbiota at a systems level. Using real-time quantitative polymerase chain reaction (PCR), Cani and colleagues [93] measured gut bacterial populations in ob/ob mice which also exhibited impaired gut permeability. They demonstrated an association between systemic metabolic dysfunction (including endotoxemia and inflammation) and alternations in the abundances of Bifidobacterium, Lactobacillus and Clostridium coccoides-Eubacterium rectale cluster. Importantly, this study identified the relationship between Clostridia and metabolic dysregulation, which has not been previously noted in monocolonisation studies. The 55role of Clostridia in gut inflammation and barrier function is further substantiated by metagenomic analysis which examines the genomic profile of the entire gut microbial community. Metagenomic studies of gut microbiota showed that patients with inflammatory bowel disease had a lower relative abundance of Clostridial cluster IV and XIVa as compared to healthy controls [94]. This suggests that the absence of these Clostridial clusters may enhance gut permeability and subsequently increase host susceptibility to chronic inflammation. There is also some evidence suggesting that bacteria in Clostridial cluster IV and XIVa are potent inducers of gut regulatory CD4 T cells, which are important modulators in the initiation of immune responses [95].
In summary, numerous studies revealed the role of certain gut microbes in modulating intestinal permeability. The effect of bacteria on gut health appears to be highly species- and even strain-specific. Identifying beneficial strains will be important for developing nutraceutical, and even pharmaceutical, interventions to improve gut health.
Gut microbiota and energy homeostasis
Gut microbiota influence host energy metabolism by modulating nutrient absorption and energy storage. There is some evidence suggesting that gut bacteria stimulate angiogenesis in the small intestine epithelium and therefore increase the efficiency of nutrient absorption [96]. It is also well documented that gut microbiota ferments dietary compounds, which are otherwise indigestible by the host, and therefore increases energy harvest.
The effect of gut microbiota on host energy homeostasis is primarily a consequence of short-chain fatty acids (SCFAs) production. Bacterial enzymes, eg glycoside hydrolases, break down dietary polysaccharides to SCFAs such as butyrate, acetate and propionate [97]. While butyrate is the primary energy substrate for colonocytes and is important for fortification of the GIT epithelial barrier, acetate and propionate are delivered to the liver for de novo lipogenesis through acetyl-CoA carboxylase and fatty acid synthase. The direct effect of gut microbiota on hepatic lipid metabolism is demonstrated in conventionalisation studies, in which the colonisation of germ-free mice with cecal content of conventionally raised animals increased fatty acid and triglyceride synthesis in the liver and promoted peripheral fat storage [98]. Bacterial SCFAs may also directly modulate the signalling pathways involved in host fat storage. SCFAs are specific ligands for at least two G protein coupled receptors, GPR 41 and GPR 43, which when deficient ameliorate microbe-associated energy harvest [99] and diet-induced obesity [100].
It is important to note that the interactions between gut microbiota, GIT and liver are part of the normal physiological processes in the host. Disturbances or alterations to the microbial community (collectively known as microbial dysbiosis) however, are likely to shift the energy balance in favour of nutrient recovery and storage. This notion is best illustrated in a series of studies by Turnbaugh, Gordon and colleagues. Germ-free mice receiving an obesity-associated microbiota (OAM, from diet-induced obese mice) had increased fat deposition as compared to those transplanted with a lean-associated microbiota (LAM) [101]. Further, it has also been shown that OAM is enriched for genes 56that encode enzymes involved in starch, sucrose, and galactose metabolism to breakdown otherwise indigestible polysaccharides [102]. These data suggest that OAM has a higher energy harvesting potential. The increased influx of SCFAs into the systemic and, more importantly, the portal circulation may increases lipid load in the liver and predispose hepatic insulin resistance.
The research of gut microbiota currently focuses on unravelling microbial populations affected in microbial dysbiosis and the associated metabolic sequelae. A feature of many human [103] and experimental models of obesity [104], is that an OAM is characterised by a lower Bacteroidetes:Firmicutes ratio as compared to an LAM. Whether this is a generic trend across the obesity-associated metabolic disorders is not entirely clear. For example, patients with type 2 diabetes have been shown to have similar Bacteroidetes:Firmicutes ratio as healthy controls but the proportion of bacteria represented within Bacteroidetes differed in the two cohorts [105]. To date there is limited evidence suggesting the predominant role of a particular microbe or a specific group of bacterial species in the events leading up to metabolic disorders. Experimental data however, strongly indicate that the composition of gut microbiota is an important aspect of host metabolic phenotype. Microbial dysbiosis, therefore, should be considered as an additional risk factor in the pathogenesis of insulin resistance and systemic metabolic dysregulations.
Future directions: focus on immune and gut systems
The discovery of the involvement of the immune and gut systems in obesity-related metabolic dysfunctions identify a subset of at-risk individuals who would benefit from novel immune- and gut-targeted therapies to improve metabolic health. Here we highlight some recent data to identify potential therapeutic targets.
Immunomodulators
A logical approach to prevent inflammation-associated metabolic sequelae is to block the initiation of an immune response. The chronic use of agents which non-selectively antagonise the key pro-inflammatory pathways (eg glucocorticoids), however, are often associated with immunosuppression-related side effects [106]. Attempts to develop interventions to reduce localised inflammation have also proven to be impractical. In the gut system, inhibiting the signalling of specific TLRs interferes with mucosal repair [107] and has even been shown to induce ‘hallmark features of metabolic syndrome’ including insulin resistance, hyperlipidemia and increased adiposity [108, 109]. It then becomes apparent that a specific TLR functional deficiency is compensated by the activation of other TLRs [108]. Further, this feedback loop appears to modulate gut microbiota profile [109] and therefore may subsequently lead to metabolically deleterious phenotypes.
Promoting resolution has recently been appreciated as an alternative way to minimise the deleterious effects of inflammation. Rather than directly interfering with the inflammatory signalling pathways, pro-resolving mediators reduce the infiltration and, at the same time, enhance the clearance of immune cells at the site of inflammation [106]. Accordingly, these mediators promote tissue recovery and therefore prevent unnecessarily prolonged 57inflammation. Resolvins are a family of endogenous pro-resolution molecules which have received much of the attention. N-3 polyunsaturated fatty acids have long been recognised as anti-inflammatory due to the preferential production of less inflammatory eicosanoids [110]. The recent discovery of the D- and E-series of resolvins, derived from docosahexaenoic acid (DHA; 22:6n-3) and eicosapentaenoic acid (EPA; 20:5n-3) respectively, suggests that the pro-resolving nature is another important aspect of n-3 polyunsaturated fatty acids to modulate inflammation. Human clinical trials of resolvins to treat inflammatory diseases including rheumatoid arthritis and inflammatory bowel disease are already underway. Consistent with the further role of inflammation in obesity/diabetes, resolvin D1 administration has recently been shown to reduce CLS-localised ATMs in visceral adipose tissue and improve insulin sensitivity in db/db mice [111]. Taken with the protective effect of fish oil feeding against LPS-induced inflammation and insulin resistance [112], these results also point to the potential of resolvins as a pharmacological target for obesity and diabetes.
Probiotics, prebiotics and resistant starches
The compelling evidence of the role of gut microbiota in gut functions and energy metabolism clearly indicates manipulating the microbial community as an important avenue to improve metabolic health. Probiotic supplementation, which involves the ingestion of live micro-organisms, is the most direct way to introduce specific beneficial bacteria into the gut system. Strains of several species of Lactobacillus and Bifidobacteria, eg L. plantarum [113] and B. bifidum [114] are probiotics with consistently demonstrated health benefits. There is an emerging literature supporting the use of probiotics supplementary to standard treatment for inflammatory bowel disease [115] and irritable bowel syndrome [116]. Dietary probiotic supplements and food fortified with probiotics (eg dairy products and infant formulas) are also widely available for general consumption. There have been concerns however, about the efficacy of probiotic supplementation as the effective dose of beneficial bacteria reaching the GIT may be highly variable and it is likely to account for only a relatively small proportion of the entire microbial population. Also little is known about the duration of effect so dosage may be critical for long-term health benefits.
Supplementation of prebiotics in combination with resistant starch is an alternative way to manipulate gut microbiota profile. Prebiotics are oligosaccharides which serve as substrates for specific gut microbes. For example inulin is a fructan preferentially used by Lactobacillus and Bifidobacteria [117]. Resistant starch is defined as starch and/or products of starch degradation, which are not absorbed in the small intestine, and therefore enters the colon with butyrate as a predominant product from microbial fermentation [118]. While each bacterial genus or species has its own preferential substrates, prebiotics and resistant starch promote the growth of specific beneficial bacterial populations and subsequently shifts the balance of microbial communities in a way that favours gut and metabolic health.
The use of food ingredients to manipulate gut microbiota composition is advantageous to probiotics supplementation. Bioavailability becomes less of an issue, but perhaps what makes prebiotics and resistant starch a really appealing option is their ability to modify, 58long term, autochthonous microbial communities and therefore increase the likelihood of having persisting health benefits. There is also the possibility of engineering dietary components to facilitate colonisation of specific microbial populations and/or to produce specific species of SCFAs to serve particular therapeutic purposes. Rats fed with diet containing 10% butyrylated high-amylose maize starch, for example, had increased total SCFAs and in particular butyrate content in the colon as compared to those which consumed non-butyrylated carbohydrates [119]. Finally, the notion of synbiotics (a combination of probiotics and prebiotics), which potentially introduce and, at the same time, maintain beneficial microbes in the gut system, may well be the most promising intervention to modify gut microbiota profile and achieve maximal health benefits.
Gut mucosal defence
Strengthening the innate defence mechanisms against pathogens is critical to maintain gut health. The gastrointestinal tract is coated with a mucus layer, as the first line of defence, to protect the epithelium from both physical and chemical damage. Mucins, the major component of the overlaying mucus layer, are glycoproteins produced primarily by goblet cells. The highly complex oligosaccharide side-chains of mucins form a viscous lining which interacts with and trap microbes and subsequently prevent direct contact of epithelial surface with pathogens [120]. The interaction between mucins and bacteria has also been shown to facilitate specific patterns of bacterial colonisation [134]. A study by An et al. [121] provided direct evidence for the role of mucins in gut function, in which mice deficient in the biosynthesis of core 3 O-glycans (the predominant component of mucins) had increased gut permeability and were more susceptible to experimental colitis and colorectal adenocarcinoma. Similarly, mice deficient in Muc2 (the most abundant mucin) exhibited signs of spontaneous colitis and growth retardation [122].
Dietary supplementations to induce mucins expression may be important to ameliorate metabolic sequelae associated with gut inflammation. Probiotics (eg specific strains of Lactobacillus) have been shown to increase mRNA levels of mucins in colonic cells in vitro [123, 124]. There is also some evidence for the ability of probiotic administration to induce gene expression of mucins in animal models of colitis [125]. Similarly, dietary supplementation of amino acids specific to mucins production restores the colonic protein level of mucins to that in the controls and promotes epithelial repair in rats with experimental colitis [126].
Trefoil factors (TFF) are another group of important proteins involved in the maintenance of the mucosal barrier. Among this family of small peptides, TFF3 is one of the most abundant secretory products from goblet cells. TFF3 works synergistically with mucins to strengthen the structural integrity of the intestinal mucosal barrier [120]. TFF3 is also critical in aiding epithelial repair following injury by promoting epithelial restitution via the TGF-beta-dependent pathway [127]. When subjected to experimental colitis, mice deficient in TFF3 are more susceptible to mortality and exhibit delayed mucosal healing as a consequence of inhibited anti-apoptosis during acute inflammation [128].59
TFF3 serves as a critical molecular link between microbiota and intestinal integrity. Commensal bacteria activate many members of the TLR family (eg TLR2 and TLR4), which subsequently induce the expression of TFF3 via the Ras/MEK/MAPK and PI3K/Akt pathways [120]. Accordingly, TFF3 is the downstream effector of the microbiota-initiated innate immune response. Manipulating TLRs, as we have argued earlier, might be a dangerous impairment of the innate immune system. However, modulating TFF3 may offer an alternate opportunity to develop interventions to improve gut health while bypassing the upstream effects of microbiota and inflammatory and/or stress-activated pathways on epithelial function. This notion is best-illustrated in a study by Podolsky and colleagues [128], in which administration of a TLR2 agonist in TFF3-/- mice and oral supplementation of recombinant TFF3 in TLR2-/- mice both confer protection of the intestinal mucosa during experimental colitis.
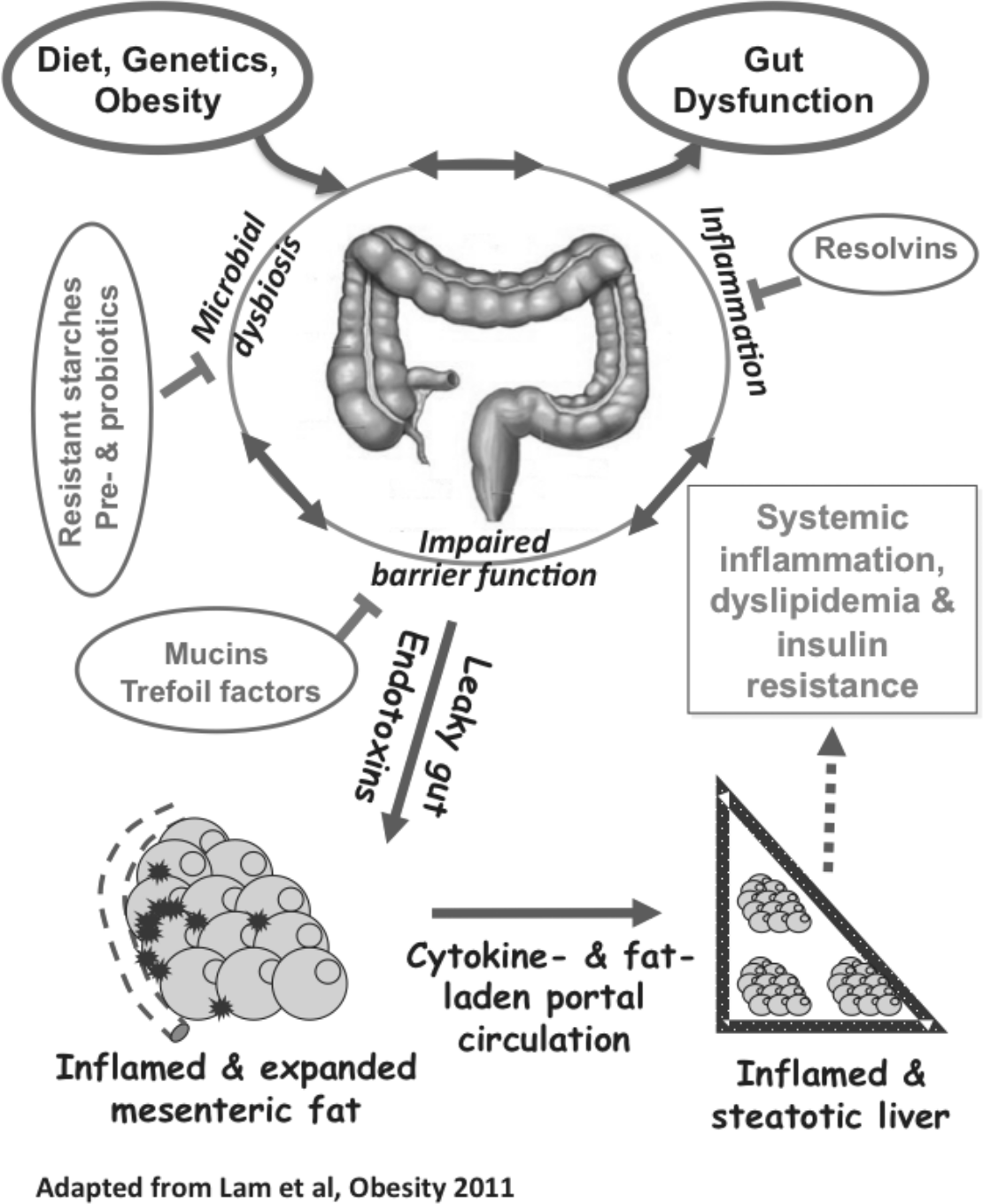
Figure 2. The role of gut in the development of systematic inflammation and metabolic dysfunctions. 60
Conclusion
Insulin resistance is central to obesity-associated metabolic dysfunctions. The little success we have in reversing the insulin-resistant state clearly suggest the need to focus on preventative measures to achieve maximal metabolic health. Recent advances in understanding the metabolic sequelae of visceral fat deposition and gut dysfunction, summarised in Figure 2, provide unprecedented opportunities to both prevent and treat metabolic disorders. What is critical now is to develop biomarkers for large-scale population screening to identify individuals with high metabolic risk and offer early preventative interventions. Dietary modifications via the development of fortified and functional foods, perhaps in combination with novel pharmaceuticals, are also promising avenues to improve metabolic health at the population level.
References
1. Reaven G (2005). All obese individuals are not created equal: insulin resistance is the major determinant of cardiovascular disease in overweight/obese individuals. Diabetes and Vascular Disease Research, 2(3): 105–12.
2. Rasouli N, Molavi B, Elbein SC & Kern PA (2007). Ectopic fat accumulation and metabolic syndrome. Diabetes, Obesity and Metabolism, 9(1): 1–10.
3. Meigs JB, Wilson PW, Fox CS, Vasan RS, Nathan DM, Sullivan LM, et al. (2006). Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease. The Journal of Clinical Endocrinology and Metabolism, 91(8): 2906–12.
4. Weiss R (2007). Fat distribution and storage: how much, where, and how? European Journal of Endocrinology, 157(Suppl. 1): S39–45.
5. Bjorntorp P (1990). ‘Portal’ adipose tissue as a generator of risk factors for cardiovascular disease and diabetes. Arteriosclerosis, 10(4): 493–96.
6. Wajchenberg BL (2000). Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocrine Reviews, 21(6): 697–738.
7. Ito H, Ohshima A, Ohto N, Ogasawara M, Tsuzuki M, Takao K, et al. (2001). Relation between body composition and age in healthy Japanese subjects. European Journal of Clinical Nutrition, 55(6): 462–70.
8. Piers LS, Rowley KG, Soares MJ & O’Dea K (2003). Relation of adiposity and body fat distribution to body mass index in Australians of Aboriginal and European ancestry. European Journal of Clinical Nutrition, 57(8): 956–63.
9. Casas YG, Schiller BC, DeSouza CA & Seals DR (2001). Total and regional body composition across age in healthy Hispanic and white women of similar socioeconomic status. The American Journal of Clinical Nutrition, 73(1): 13–18.
10. Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, et al. (2007). Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation, 116(1): 39–48.61
11. Kelley DE, Thaete FL, Troost F, Huwe T & Goodpaster BH (2000). Subdivisions of subcutaneous abdominal adipose tissue and insulin resistance. American Journal of Physiology – Endocrinology and Metabolism, 278(5): E941–48.
12. Cnop M, Landchild MJ, Vidal J, Havel PJ, Knowles NG, Carr DR, et al. (2002). The concurrent accumulation of intra-abdominal and subcutaneous fat explains the association between insulin resistance and plasma leptin concentrations: distinct metabolic effects of two fat compartments. Diabetes, 51(4): 1005–15.
13. Hayashi T, Boyko EJ, McNeely MJ, Leonetti DL, Kahn SE & Fujimoto WY (2008). Visceral adiposity, not abdominal subcutaneous fat area, is associated with an increase in future insulin resistance in Japanese Americans. Diabetes, 57(5): 1269–75.
14. Karelis AD, St-Pierre DH, Conus F, Rabasa-Lhoret R & Poehlman ET (2004). Metabolic and body composition factors in subgroups of obesity: what do we know? The Journal of Clinical Endocrinology and Metabolism, 89(6): 2569–75.
15. Lien LF, Haqq AM, Arlotto M, Slentz CA, Muehlbauer MJ, McMahon RL, et al. (2009). The STEDMAN project: biophysical, biochemical and metabolic effects of a behavioral weight loss intervention during weight loss, maintenance, and regain. OMICS, 13(1): 21–35.
16. Carr DB, Utzschneider KM, Hull RL, Kodama K, Retzlaff BM, Brunzell JD, et al. (2004). Intra-abdominal fat is a major determinant of the National Cholesterol Education Program Adult Treatment Panel III criteria for the metabolic syndrome. Diabetes, 53(8): 2087–94.
17. Ibanez J, Izquierdo M, Arguelles I, Forga L, Larrion JL, Garcia-Unciti M, et al. (2005). Twice-weekly progressive resistance training decreases abdominal fat and improves insulin sensitivity in older men with type 2 diabetes. Diabetes Care, 28(3): 662–67.
18. Wagenknecht LE, Langefeld CD, Scherzinger AL, Norris JM, Haffner SM, Saad MF, et al. (2003). Insulin sensitivity, insulin secretion, and abdominal fat: the Insulin Resistance Atherosclerosis Study (IRAS) Family Study. Diabetes, 52(10): 2490–96.
19. Cnop M, Landchild MJ, Vidal J, Havel PJ, Knowles NG, Carr DR, et al. (2002). The concurrent accumulation of intra-abdominal and subcutaneous fat explains the association between insulin resistance and plasma leptin concentrations: distinct metabolic effects of two fat compartments. Diabetes, 51(4): 1005–15.
20. Miyazaki Y, Glass L, Triplitt C, Wajcberg E, Mandarino LJ & DeFronzo RA (2002). Abdominal fat distribution and peripheral and hepatic insulin resistance in type 2 diabetes mellitus. American Journal of Physiology – Endocrinology and Metabolism, 283(6): E1135–43.
21. Porter SA, Massaro JM, Hoffmann U, Vasan RS, O’Donnel CJ & Fox CS (2009). Abdominal subcutaneous adipose tissue: a protective fat depot? Diabetes Care, 32(6): 1068–75.
22. Walker GE, Verti B, Marzullo P, Savia G, Mencarelli M, Zurleni F, et al. (2007). Deep subcutaneous adipose tissue: a distinct abdominal adipose depot. Obesity, 15(8): 1933–43.
23. Ross R, Freeman J, Hudson R & Janssen I (2002). Abdominal obesity, muscle composition, and insulin resistance in premenopausal women. The Journal of Clinical Endocrinology and Metabolism, 87(11): 5044–51.62
24. Ibrahim MM (2010). Subcutaneous and visceral adipose tissue: structural and functional differences. Obesity Reviews, 11(1): 11–18.
25. Rabe K, Lehrke M, Parhofer KG & Broedl UC (2008). Adipokines and insulin resistance. Molecular Medicine, 14(11–12): 741–51.
26. Gregor MF & Hotamisligil GS (2011). Inflammatory mechanisms in obesity. Annual Review of Immunology, 23(29): 415–45.
27. Crook M (2004). Type 2 diabetes mellitus: a disease of the innate immune system? An update. Diabetic Medicine, 21(3): 203–07.
28. Hu FB, Meigs JB, Li TY, Rifai N & Manson JE (2004). Inflammatory markers and risk of developing type 2 diabetes in women. Diabetes, 53(3): 693–700.
29. Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, et al. (2002). Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. The Journal of Clinical Investigation, 109(10): 1321–26.
30. Panagiotakos DB, Pitsavos C, Yannakoulia M, Chrysohoou C & Stefanadis C (2005). The implication of obesity and central fat on markers of chronic inflammation: The ATTICA study. Atherosclerosis, 183(2): 308–15.
31. Krysiak R, Labuzek K & Okopien B (2009). Effect of atorvastatin and fenofibric acid on adipokine release from visceral and subcutaneous adipose tissue of patients with mixed dyslipidemia and normolipidemic subjects. Pharmacological Reports, 61(6): 1134–45.
32. Bruun JM, Lihn AS, Madan AK, Pedersen SB, Schiott KM, Fain JN, et al. (2004). Higher production of IL-8 in visceral vs subcutaneous adipose tissue. Implication of nonadipose cells in adipose tissue. American Journal of Physiology – Endocrinology and Metabolism, 286(1): E8–13.
33. Lam YY, Janovska A, McAinch AJ, Belobrajdic DP, Hatzinikolas G, Game P, et al. (2011). The use of adipose tissue-conditioned media to demonstrate the differential effects of fat depots on insulin-stimulated glucose uptake in a skeletal muscle cell line. Obesity Research and Clinical Practice, 5: e43–e54.
34. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, et al. (2005). IKK-beta links inflammation to obesity-induced insulin resistance. Nature Medicine,11(2): 191–98.
35. Austin RL, Rune A, Bouzakri K, Zierath JR & Krook A (2008). siRNA-mediated reduction of inhibitor of nuclear factor-kappaB kinase prevents tumor necrosis factor-alpha-induced insulin resistance in human skeletal muscle. Diabetes, 57(8): 2066–73.
36. Fain JN (2006). Release of interleukins and other inflammatory cytokines by human adipose tissue is enhanced in obesity and primarily due to the nonfat cells. Vitamins and Hormones, 74: 443–77.
37. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL & Ferrante AW Jr (2003). Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation, 112(12): 1796–808.
38. Harman-Boehm I, Bluher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, et al. (2007). Macrophage infiltration into omental versus subcutaneous fat across different populations: effect 63of regional adiposity and the comorbidities of obesity. The Journal of Clinical Endocrinology and Metabolism, 92(6): 2240–47.
39. Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, et al. (2006). CCR2 modulates inflammatory and metabolic effects of high-fat feeding. The Journal of Clinical Investigation, 116(1): 115–24.
40. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. (2006). MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. The Journal of Clinical Investigation, 116(6):1494–1505.
41. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. (2005). Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. The Journal of Lipid Research, 46(11): 2347–55.
42. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, 2nd, DeFuria J, Jick Z, et al. (2007). Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes, 56(12): 2910–18.
43. Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. (2008). Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. The Journal of Lipid Research, 49(7): 1562–68.
44. Spencer M, Yao-Borengasser A, Unal R, Rasouli N, Gurley CM, Zhu B, et al. (2010). Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. American Journal of Physiology – Endocrinology and Metabolism, 99(6): E1016–27.
45. Pasarica M, Gowronska-Kozak B, Burk D, Remedios I, Hymel D, Gimble J, et al. (2009). Adipose tissue collagen VI in obesity. The Journal of Clinical Endocrinology and Metabolism, 94(12): 5155–62.
46. Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, et al. (2009). Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Molecular and Cellular Biology, 29(6): 1575–91.
47. Gregor MF & Hotamisligil GS (2007). Thematic review series: Adipocyte Biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. The Journal of Lipid Research, 48(9): 1905–14.
48. Zhang K & Kaufman RJ (2008). From endoplasmic-reticulum stress to the inflammatory response. Nature, 454(7203): 455–62.
49. de Ferranti S & Mozaffarian D (2008). The perfect storm: obesity, adipocyte dysfunction, and metabolic consequences. Clinical Chemistry, 54(6): 945–55.
50. Martinez FO, Sica A, Mantovani A & Locati M (2008). Macrophage activation and polarization. Frontiers in Bioscience, 13: 453–61.
51. Lumeng CN, DelProposto JB, Westcott DJ & Saltiel AR (2008). Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes, 57(12): 3239–46.
52. Aron-Wisnewsky J, Tordjman J, Poitou C, Darakhshan F, Hugol D, Basdevant A, et al. (2009). Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. The Journal of Clinical Endocrinology and Metabolism, 94(11): 4619–23.64
53. Shaul ME, Bennett G, Strissel KJ, Greenberg AS & Obin MS (2010). Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet-induced obesity in mice. Diabetes, 59(5): 1171–81.
54. Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre-Bosquet N, et al. (2005). Macrophage activation switching: an asset for the resolution of inflammation. Clinical and Experimental Immunology, 142(3): 481–89.
55. Desreumaux P, Ernst O, Geboes K, Gambiez L, Berrebi D, Muller-Alouf H, et al. (1999). Inflammatory alterations in mesenteric adipose tissue in Crohn’s disease. Gastroenterology, 117(1): 73–81.
56. Maconi G, Greco S, Duca P, Ardizzone S, Massari A, Cassinotti A, et al. (2008). Prevalence and clinical significance of sonographic evidence of mesenteric fat alterations in Crohn’s disease. Inflammatory Bowel Diseases, 14(11): 1555–61.
57. Bertin B, Desreumaux P & Dubuquoy L (2010). Obesity, visceral fat and Crohn’s disease. Current Opinion in Clinical Nutrition and Metabolic Care, 13(5): 574–80.
58. Gambero A, Marostica M, Abdalla Saad MJ & Pedrazzoli J Jr (2007). Mesenteric adipose tissue alterations resulting from experimental reactivated colitis. Inflammatory Bowel Diseases, 13(11): 1357–64.
59. Thomaz MA, Acedo SC, de Oliveira CC, Pereira JA, Priolli DG, Saad MJ, et al. (2009). Methotrexate is effective in reactivated colitis and reduces inflammatory alterations in mesenteric adipose tissue during intestinal inflammation. Pharmacological Research, 60(4): 341–46.
60. Cereijido M, Contreras RG, Flores-Benitez D, Flores-Maldonado C, Larre I, Ruiz A, et al. (2007). New diseases derived or associated with the tight junction. Archives of Medical Research, 38(5): 465–78.
61. Muller CA, Autenrieth IB & Peschel A (2005). Innate defenses of the intestinal epithelial barrier. Cellular and Molecular Life Sciences, 62(12): 1297–307.
62. D’Inca R, Annese V, di Leo V, Latiano A, Quaino V, Abazia C, et al. (2006). Increased intestinal permeability and NOD2 variants in familial and sporadic Crohn’s disease. Alimentary Pharmacology and Therapeutics, 23(10): 1455–61.
63. Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, et al. (2002). Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. American Journal of Pathology, 161(5): 1903–15.
64. Ye D, Ma I & Ma TY (2006). Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. American Journal of Physiology Gastrointestinal and Liver Physiology, 290(3): G496–504.
65. Al-Sadi R, Ye D, Dokladny K & Ma TY (2008). Mechanism of IL-1beta-induced increase in intestinal epithelial tight junction permeability. The Journal of Immunology, 180(8): 5653–61.
66. Bumrungpert A, Kalpravidh RW, Chitchumroonchokchai C, Chuang CC, West T, Kennedy A, et al. (2009). Xanthones from mangosteen prevent lipopolysaccharide-mediated inflammation and insulin resistance in primary cultures of human adipocytes. Journal of Nutrition, 139(6): 1185–91.65
67. Grisouard J, Bouillet E, Timper K, Radimerski T, Dembinski K, Frey DM, et al. (2010). Both inflammatory and classical lipolytic pathways are involved in lipopolysaccharides-induced lipolysis in human adipocytes. Innate Immunity, 18 November, DOI: 10.1177/1753425910386632 [Online]. Available: ini.sagepub.com/content/early/2010/10/14/1753425910386632.full.pdf [Accessed 12 January 2012].
68. Kopp A, Bala M, Buechler C, Falk W, Gross P, Neumeier M, et al. (2010). C1q/TNF-related protein-3 represents a novel and endogenous lipopolysaccharide antagonist of the adipose tissue. Endocrinology, 151(11): 5267–78.
69. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A & Locati M (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology, 25(12): 677–86.
70. Pond CM (2005). Adipose tissue and the immune system. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 73(1): 17–30.
71. Peyrin-Biroulet L, Chamaillard M, Gonzalez F, Beclin E, Decourcelle C, Antunes L, et al. (2007). Mesenteric fat in Crohn’s disease: a pathogenetic hallmark or an innocent bystander? Gut, 56(4): 577–83.
72. Tarantino G, Savastano S & Colao A (2010). Hepatic steatosis, low-grade chronic inflammation and hormone/growth factor/adipokine imbalance. World Journal of Gastroenterology, 16(38): 4773–83.
73. Postic C, Dentin R & Girard J (2004). Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes and Metabolism, 30(5): 398–408.
74. Szabo G & Bala S (2010). Alcoholic liver disease and the gut-liver axis. World Journal of Gastroenterology, 16(11): 1321–29.
75. Rao R (2009). Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology, 50(2): 638–44.
76. Pastor Rojo O, Lopez San Roman A, Albeniz Arbizu E, de la Hera Martinez A, Ripoll Sevillano E & Albillos Martinez A (2007). Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflammatory Bowel Diseases, 13(3): 269–77.
77. Sun L, Yu Z, Ye X, Zou S, Li H, Yu D, et al. (2010). A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care, 8 June, 33(9):1925-32. DOI: 10.2337/dc10-0340 [Online]. Available: care.diabetesjournals.org/content/early/2010/06/03/dc10-0340.full.pdf+html [Accessed 12 January 2012].
78. Creely SJ, McTernan PG, Kusminski CM, Fisher M, Da Silva NF, Khanolkar M, et al. (2007). Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. American Journal of Physiology – Endocrinology and Metabolism, 292(3): E740–47.
79. Manco M, Putignani L & Bottazzo GF (2010). Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocrine Reviews, 31(6): 817–44.
80. Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, et al. (2010). Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes, 59(1): 172–81.66
81. Lu YC, Yeh WC & Ohashi PS (2008). LPS/TLR4 signal transduction pathway. Cytokine, 42(2): 145–51.
82. Kim JJ & Sears DD (2010). TLR4 and insulin resistance. Gastroenterology Research and Practice, 2010.
83. Savage DC (1977). Microbial ecology of the gastrointestinal tract. Annual Review of Microbiology, 31: 107–33.
84. Xu J & Gordon JI. (2003). Honor thy symbionts. Proceedings of the National Academy of Sciences of the United States of America, 100(18): 10452–59.
85. Camp JG, Kanther M, Semova I & Rawls JF (1989). Patterns and scales in gastrointestinal microbial ecology. Gastroenterology, 136(6): 1989–2002.
86. Bocci V (1992). The neglected organ: bacterial flora has a crucial immunostimulatory role. Perspectives in Biology and Medicine, 35(2): 251–60.
87. Macfarlane S, Woodmansey EJ & Macfarlane GT (2005). Colonization of mucin by human intestinal bacteria and establishment of biofilm communities in a two-stage continuous culture system. Applied and Environmental Microbiology, 71(11): 7483–92.
88. Anderson RC, Cookson AL, McNabb WC, Park Z, McCann MJ, Kelly WJ, et al. (2010). Lactobacillus plantarum MB452 enhances the function of the intestinal barrier by increasing the expression levels of genes involved in tight junction formation. BMC Microbiology, 10: 316.
89. Karczewski J, Troost FJ, Konings I, Dekker J, Kleerebezem M, Brummer RJ, et al. (2010). Regulation of human epithelial tight junction proteins by Lactobacillus plantarum in vivo and protective effects on the epithelial barrier. American Journal of Physiology Gastrointestinal and Liver Physiology, 298(6): G851–59.
90. Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, et al. (2011). The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metabolism, 13(5): 517–26.
91. Bernet-Camard MF, Lievin V, Brassart D, Neeser JR, Servin AL & Hudault S (1997). The human Lactobacillus acidophilus strain LA1 secretes a nonbacteriocin antibacterial substance(s) active in vitro and in vivo. Applied and Environmental Microbiology, 63(7): 2747–53.
92. Ewaschuk JB, Diaz H, Meddings L, Diederichs B, Dmytrash A, Backer J, et al. (2008). Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. American Journal of Physiology Gastrointestinal and Liver Physiology, 295(5): G1025–34.
93. Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, et al. (2009). Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut, 58(8): 1091–103.
94. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N & Pace NR (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America, 104(34): 13780–85.67
95. Smith PM & Garrett WS (2011). The gut microbiota and mucosal T cells. Frontiers in Microbiology, 2: 111.
96. Stappenbeck TS, Hooper LV & Gordon JI (2002). Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proceedings of the National Academy of Sciences of the United States of America, 99(24): 15451–55.
97. Flint HJ, Duncan SH, Scott KP & Louis P (2007). Interactions and competition within the microbial community of the human colon: links between diet and health. Environmental Microbiology, 9(5): 1101–11.
98. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proceedings of the National Academy of Sciences of the United States of America, 101(44): 15718–23.
99. Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, et al. (2008). Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proceedings of the National Academy of Sciences of the United States of America, 105(43): 16767–72.
100. Bjursell M, Admyre T, Goransson M, Marley AE, Smith DM, Oscarsson J, et al. (2011). Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. American Journal of Physiology – Endocrinology and Metabolism, 300(1): E211–20.
101. Turnbaugh PJ, Backhed F, Fulton L & Gordon JI (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host and Microbe, 3(4): 213–23.
102. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER & Gordon JI (2006). An obesityassociated gut microbiome with increased capacity for energy harvest. Nature, 444(7122): 1027–31.
103. Ley RE, Turnbaugh PJ, Klein S & Gordon JI. (2006). Microbial ecology: human gut microbes associated with obesity. Nature, 444(7122): 1022–23.
104. Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD & Gordon JI (2005). Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America, 102(31): 11070–75.
105. Wu X, Ma C, Han L, Nawaz M, Gao F, Zhang X, et al. (2010). Molecular characterisation of the faecal microbiota in patients with type II diabetes. Current Microbiology, 61(1): 69–78.
106. Uddin M & Levy BD (2011). Resolvins: natural agonists for resolution of pulmonary inflammation. Progress in Lipid Research, 50(1): 75–88.
107. Ungaro R, Fukata M, Hsu D, Hernandez Y, Breglio K, Chen A, et al. (2009). A novel toll-like receptor 4 antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. American Journal of Physiology Gastrointestinal and Liver Physiology, 296(6): G1167–79.
108. Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, et al. (2007). Deletion of TLR5 results in spontaneous colitis in mice. The Journal of Clinical Investigation, 117(12): 3909–21.
109. Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. (2010). 68Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science, 328(5975): 228–31.
110. Adkins Y & Kelley DS (2010). Mechanisms underlying the cardioprotective effects of omega-3 polyunsaturated fatty acids. The Journal of Nutritional Biochemistry, 21(9): 781–92.
111. Hellmann J, Tang Y, Kosuri M, Bhatnagar A & Spite M (2011). Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. The FASEB Journal, 25: 2399–407.
112. Vijay-Kumar M, Vanegas SM, Patel N, Aitken JD, Ziegler TR, Ganji V. (2011). Fish oil rich diet in comparison to saturated fat rich diet offered protection against lipopolysaccharide-induced inflammation and insulin resistance in mice. Nutrition and Metabolism, 8(1): 16.
113. Molin G (2001). Probiotics in foods not containing milk or milk constituents, with special reference to Lactobacillus plantarum 299v. The American Journal of Clinical Nutrition, 73(2): 380S–5S.
114. Trebichavsky I, Rada V, Splichalova A & Splichal I (2009). Cross-talk of human gut with bifidobacteria. Nutrition Reviews, 67(2): 77–82.
115. Cary VA & Boullata J (2010). What is the evidence for the use of probiotics in the treatment of inflammatory bowel disease? Journal of Clinical Nursing, 19(7–8): 904–16.
116. Lee BJ & Bak YT (2011). Irritable bowel syndrome, gut microbiota and probiotics. Journal of Neurogastroenterology and Motility, 17(3): 252–66.
117. Gourbeyre P, Denery S & Bodinier M (2011). Probiotics, prebiotics, and synbiotics: impact on the gut immune system and allergic reactions. Journal of Leukocyte Biology, 89(5): 685–95.
118. Topping DL, Fukushima M & Bird AR (2003). Resistant starch as a prebiotic and synbiotic: state of the art. The Proceedings of the Nutrition Society, 62(1): 171–76.
119. Clarke JM, Topping DL, Bird AR, Young GP & Cobiac L (2008). Effects of high-amylose maize starch and butyrylated high-amylose maize starch on azoxymethane-induced intestinal cancer in rats. Carcinogenesis, 29(11): 2190–94.
120. Kim YS & Ho SB (2010). Intestinal goblet cells and mucins in health and disease: recent insights and progress. Current Gastroenterology Reports, 12(5): 319–30.
121. An G, Wei B, Xia B, McDaniel JM, Ju T, Cummings RD, et al. (2007). Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. The Journal of Experimental Medicine, 204(6): 1417–29.
122. Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. (2006). Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology, 131(1): 117–29.
123. Mack DR, Michail S, Wei S, McDougall L & Hollingsworth MA (1999). Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. American Journal of Physiology, 276(4 Pt 1): G941–50.
124. Mattar AF, Teitelbaum DH, Drongowski RA, Yongyi F, Harmon CM & Coran AG (2002). 69Probiotics up-regulate MUC-2 mucin gene expression in a Caco-2 cell-culture model. Pediatric Surgery International, 18(7): 586–90.
125. Amit-Romach E, Uni Z & Reifen R (2010). Multistep mechanism of probiotic bacterium, the effect on innate immune system. Molecular Nutrition and Food Research, 54(2): 277–84.
126. Faure M, Mettraux C, Moennoz D, Godin JP, Vuichoud J, Rochat F, et al. (2006). Specific amino acids increase mucin synthesis and microbiota in dextran sulfate sodium-treated rats. Journal of Nutrition, 136(6): 1558–64.
127. Sturm A & Dignass AU (2008). Epithelial restitution and wound healing in inflammatory bowel disease. World Journal of Gastroenterology, 14(3): 348–53.
128. Podolsky DK, Gerken G, Eyking A & Cario E (2009). Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology, 137(1): 209–20.