5
Pancreatic beta-cell failure in the pathogenesis of type 2 diabetes
As the worldwide prevalence of type 2 diabetes continues to rise, there is mounting evidence that failure of the cells which make insulin – termed pancreatic beta (or β) cells, play a fundamental role at all stages in the pathogenesis. Beta-cell defects such as loss of the initial (first) phase of insulin secretion and inefficient insulin synthesis are seen years prior to the onset of type 2 diabetes. Later, it is the failure of beta cells to adequately respond to the demand for high insulin requirements that determines the onset of hyperglycaemia leading to the clinical diagnosis of type 2 diabetes. In this chapter, we will discuss the various proposed mechanisms of beta-cell failure in type 2 diabetes and address recent controversies in this field of research.
The prevalence of type 2 diabetes is rising at an alarming rate. In Australia alone, those affected by type 2 diabetes doubled from 3.4% in 1981 to 7.4% in a national sample of 11 247 adults in 1999 to 2002. An additional 16.4% of participants showed symptoms of prediabetes in this study, a sign of imminent trouble.
On a global scale, the International Diabetes Foundation projected that by 2030, up to 438 million people may be affected by this disease, accounting for more than 4.5% of the world’s projected population [3]. China, the world’s most populous nation, has not been spared as a recent survey reported a remarkably high diabetes prevalence of 9.7% (92.4 million adults) [4].
The burden of diabetes spans beyond the threat it poses to the individual sufferer and includes the greater effect on communities, the health economy and workforce. The estimated financial cost of type 2 diabetes in Australia is A$10.3 billion per annum which includes costs for carers (>A$4 billion), productivity losses (A$4.1 billion), direct costs to the healthcare system and the estimated direct costs of obesity (A$2.2 billion) [5].
Modern life is largely to blame for this surge in diabetes prevalence with the abundance of calorie-rich foods, the gross reduction in energy expenditure resulting from the use
88of cars and other labour-saving devices, the hours spent sitting in front of television or computer screens and the avoidance of outdoor activities. As described in earlier chapters, it is the collusion of such environmental factors with an individual’s genetic predisposition that lead to pancreatic beta-cell failure that is characteristic of type 2 diabetes. However, the precise pathologic processes by which beta cells fail in their essential role in glucose homeostasis are incompletely understood and contentious.
This chapter will outline some of the proposed mechanisms of beta-cell failure in type 2 diabetes and touch on a couple of controversial issues that have characterised this field of research. However, it will first lay the groundwork with some basic principles in beta-cell physiology.
The beta cell
Since the initial description of the pancreatic islet by Paul Langerhans in 1869 as ‘a network of small cells, almost perfect in homogeneity and structure, arranged in groups, scattered amongst the acinar cells of the pancreas’ [6], understanding of this highly complex miniorgan has increased greatly. Five different endocrine cell types are found within the pancreatic islet, namely alpha cells that make glucagon, beta cells that make insulin, delta cells that make somatostatin, PP cells that make pancreatic polypeptide and epsilon cells that make ghrelin [7].
Although islets cells account for only 1%–2% of the pancreatic mass [8, 9], they receive 10% of the total pancreatic blood flow and are surrounded by a highly specialised system of fenestrated endothelial cells which provide critical signals for cell differentiation, development and function [10]. Other extracellular components, including extracellular matrix and neurons also play critical roles in influencing the behaviour of islet cells [11]. Beta cells are the dominant cell type of the human islet, accounting for 50% to 70% of islet cells [12]. Their main function, namely regulated insulin secretion, involves the integration of several stimuli which activate secretion when challenged and others that switch it off when the stimuli are no longer present. Whilst there is general agreement that the entry of glucose into the beta cell via specialised glucose transporters leads to a cascade of events including glucose phosphorylation by glucokinase and the closure of potassium-ion channels leading to membrane depolarisation and insulin secretion [13], the process is more complex. Following glucose entry into beta cells, insulin is secreted in two distinct phases and in an oscillatory, rather than a linear, pattern [14]. Further complexity can be found in the presence of secondary pathways of insulin secretion independent of ion channels [11], interactions between beta cells which influence behaviour [15] and the effect of gut-derived hormones, known as incretins, on beta-cell function [16]. The major steps in glucose-stimulated insulin secretion have been summarised in Figure 1.
However our understanding of beta-cell physiology is incomplete, with lack of understanding of the roles of a broad system of G-protein coupled receptors on the beta-cell surface [17], the effect of other nutrients such as amino acids and fatty acids on beta-cell function [18] and the role of vitamin D activating enzymes and receptors within the beta cell [19]. In 89seeking answers to these and many other questions, researchers in this field face a number of challenges. Significant inter-species differences in islet architecture [20] and responses to hyperglycaemia [21] pose a challenge that has made direct extrapolation of disease-related pathophysiology from rodents to humans tricky. Furthermore, the study of human pancreatic specimens, whilst insightful, does not always reflect in vivo beta-cell behaviour.
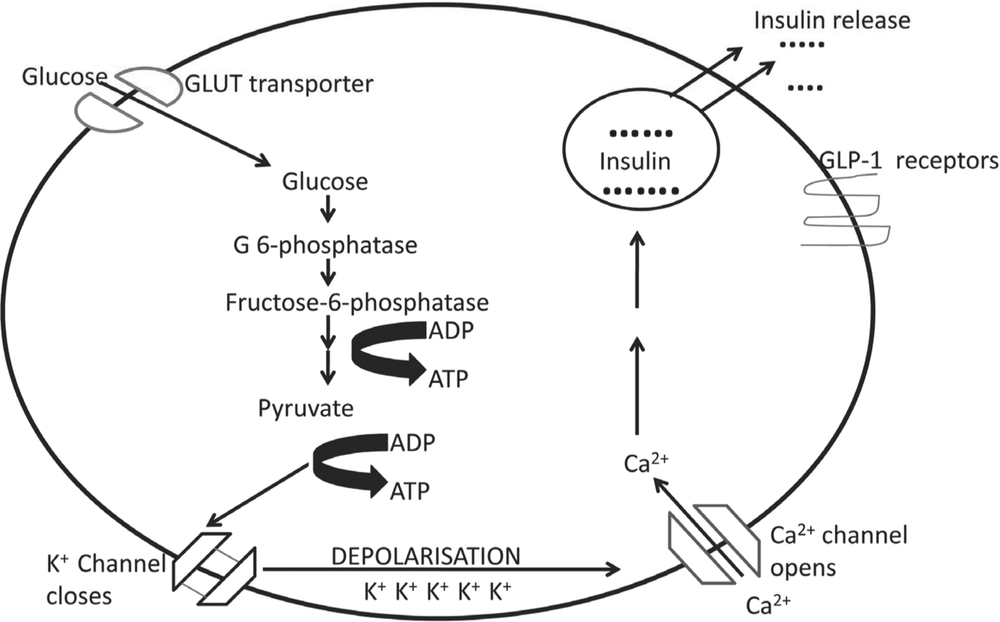
Figure 1. Steps involved in glucose-stimulated insulin secretion by the pancreatic beta cell. Following entry of glucose via GLUT transporters, phosphorylation by glucokinase occurs and pyruvate is formed via the process of glycolysis. This leads to increased activity of the TCA cycle within the mitochondria by which adenosine diphosphate (ADP) is converted to adenosine triphosphate (ATP). This causes closure of ATP-sensitive potassium channels, a wave of membrane depolarisation and the subsequent activation of voltage gated calcium channels. The entry of calcium into the cell is followed by vesicle docking and insulin granules are released into the circulation. Other factors play a role such as glucagon-like peptide 1 which has unique receptors in the beta-cell membrane.
There is no doubt that beta cells play an important role in the pathogenesis of type 2 diabetes but the complex nature of these cells and the difficulties encountered in ‘unlocking’ some of these complexities have inevitably led to some controversy in our understanding of the precise nature of this role.90
Insulin resistance or defective beta cells?
A controversial issue for many years has been whether insulin resistance or impaired beta-cell function is the primary cause of type 2 diabetes. This controversy seems to have resulted from the complex interaction between both defects present early in the disease and the relative ease in measuring in vivo insulin resistance as compared to beta-cell function [22].
A 1992 study examining non-diabetic subjects at high risk of developing type 2 diabetes (ie women with prior gestational diabetes, high risk ethnic groups, people whose parents were both affected by type 2 diabetes) reported that insulin resistance was the predominant feature [23]. This was based on a glucose tolerance test which demonstrated supraphysiological insulin levels required to maintain normal glucose at two hours following a 75 gm oral glucose load. However, by only obtaining two-hour measurements, the high insulin levels were misinterpreted as representing normal beta-cell function compensating for a state of insulin resistance. As discussed above, insulin secretion is normally biphasic and the first phase of insulin secretion, maximal at 30 minutes following a meal, appears to be the first casualty of the beta cell in type 2 diabetes [12] Therefore, if values were measured at 30 minutes following the glucose load, different levels may have been found which would match the early rise in glucose, then followed by compensatory insulin hypersecretion at two hours.
A prospective study of 200 non-diabetic Pima Indians, which is a group with a very high risk of type 2 diabetes, also reported that insulin resistance was the earliest feature in the 38 patients who went on to develop diabetes [24]. Lifestyle factors, such as obesity, sedentary lifestyle and high-fat diets correlated closely with this state of insulin resistance which was detected by the gold-standard technique, the hyperinsulinaemic-euglycaemic clamp. However, as the disease developed, the degree of insulin resistance did not change significantly but rather, a dynamic pattern of beta-cell activity emerged with early hyperinsulinaemia to maintain normoglycaemia being followed by a significant drop in insulin levels resulting in hyperglycaemia. It was therefore proposed that beta-cell failure was the final determinant in the development of hyperglycaemia in these patients.
For some time, ‘beta-cell exhaustion’ was the sole explanation for the failure in insulin secretion [20]. This is the concept that after a prolonged period of producing high amounts of insulin in order to overcome an insulin resistant state, otherwise normal beta cells eventually ‘burn out’ and permanently fail. Proponents of this hypothesis also describe distinct stages in the evolution of beta cells from a state of compensation to severe decompensation [25]. But the exhaustion concept has some limitations. Most patients with conditions characterised by insulin resistance – such as pregnancy, morbid obesity, Cushing’s disease and acromegally – do not develop diabetes. Furthermore, studies in humans and rhesus monkeys suggest that the early hyperinsulinaemia may occur independently of insulin resistance and rather reflect ‘beta-cell hyper-responsiveness’ [26]. In addition, patients with mutations in the insulin receptor, who have severe insulin resistance from birth do not develop diabetes until their beta cells fail, usually in their teens or 20s [27]. Therefore, intrinsic defects in susceptible beta cells appear to be important 91both early in the development of diabetes and later when the cells undergo critical failure in their compensatory capacity.
Another five-year prospective study involving non-diabetic Pima Indians confirmed dual pathology involving both the beta cell and peripheral insulin sensitivity in the development of diabetes [28]. Amongst 48 non-diabetic subjects who were roughly equal for parameters of obesity and insulin resistance, the 17 who progressed to diabetes failed to increase their insulin secretion in response to progression of insulin resistance. Furthermore, they showed signs of defective beta-cell function even before the onset of impaired glucose tolerance. The 31 subjects who did not progress to diabetes demonstrated appropriate increases in insulin secretion as they gained weight and became more insulin resistant. Therefore, it was beta-cell function that determined the progression from insulin resistance to frank diabetes in this high-risk group.
A useful way to understand the interaction between insulin sensitivity and beta-cell function can be found in the disposition index [29]. In normal subjects, beta-cell function varies according to the degree of insulin sensitivity so that when non-diabetic individuals enter a phase of greater insulin resistance, such as in puberty, pregnancy or aging, beta cells ‘rise to the occasion’ and produce greater insulin as required. The relationship follows a hyperbolic pattern as seen in Figure 2, indicating a continuum of beta-cell responsiveness to variations in insulin sensitivity. The dotted, downward line in this figure represents the progression to diabetes as was displayed in the previous study [25] – while still glucose tolerant, susceptible individuals began to ‘fall below the curve’ with the inability of beta cells to respond appropriately to the degree of insulin resistance. The downward, rather than horizontal, trajectory of this dotted line indicates that deterioration in beta-cell function was of greater importance than the reduction in insulin sensitivity in the development of hyperglycaemia [20]. Therefore, in essence, type 2 diabetes may be considered a failure of beta-cell compensation.
Beta-cell failure and insulin resistance however need not be considered as separate entities. Pathways are emerging by which these otherwise disparate conditions may be linked. Striking examples exist in the insulin signalling pathway. For example, insulin receptor substrate-2 (IRS-2) promotes both cell growth, insulin secretion and plays a well-recognised role in insulin sensitivity [30]. Mice with IRS-2 mutations in their beta-cells develop diabetes. In addition, people with Akt2 mutations develop diabetes due to the combination of severe insulin resistance and impaired beta-cell dysfunction [31]. Insulin receptor mutations also associate with diabetes, both in people, and in mice. The transcription factor NF-kappaB plays an important role in both insulin resistance [32] and beta-cell behaviour [33]. Furthermore, complex processes which alter the behaviour of the mitochondria, the cell’s own ‘energy plant’, are similar in insulin-resistant tissues and defective beta cells. Therefore, insulin resistance and beta-cell dysfunction may be aetiologically related and rather than predominating over the other, appear to conspire together in the development of diabetes.92
Reduced beta-cell mass or function?
Currently, there is a debate concerning the relative roles of decreased beta-cell mass compared with decreased beta-cell function in diabetes pathogenesis [34].
Proponents of the reduced beta-cell mass argument refer to postmortem studies which demonstrate up to 40% reduction in the number of beta cells in subjects with prediabetes versus weight-matched non-diabetic individuals [35]. In addition, mutations of genes known to control beta-cell mass such as the pancreatic and duodenal homeobox 1 (Pdx-1) result in monogenic forms of diabetes, such as Maturity Onset Diabetes of the Young [36].
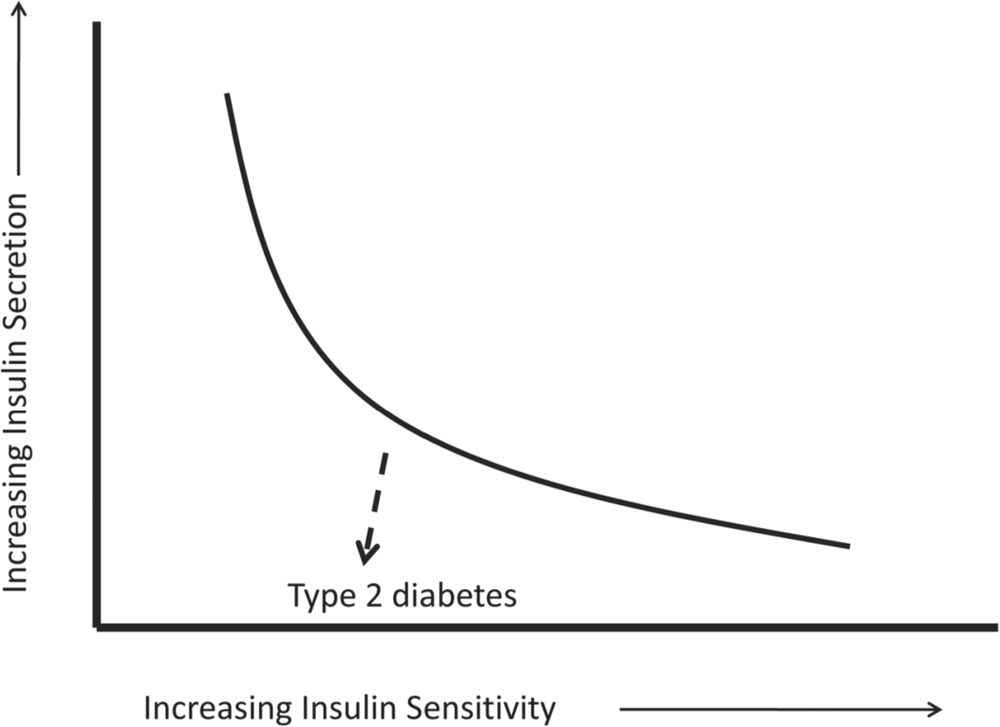
Figure 2. The ‘disposition index’ is a useful way to consider the relationship between insulin secretion and insulin sensitivity in both physiologic (ie curved line) and pathologic states (ie dotted line). This is based on data from a number of studies and has been adapted from Kahn et al. [26]. Individuals who develop type 2 diabetes head in a downward trajectory below the curve indicating that deterioration in beta-cell function (and thus less insulin secretion) is the main factor in the development of hyperglycaemia.
However, there are some limitations to this argument. Firstly, not all autopsy studies in diabetic subjects demonstrate relative reductions in beta-cell mass [37]. Secondly, beta-cell mass is highly variable even in normal individuals and may undergo substantial changes throughout a person’s life [7]. Thirdly, it is difficult to see how a reduction of less than 50% of otherwise healthy beta cells could cause diabetes in the light of a recent in vivo islet donation from 50% of a healthy human pancreas which resulted in diabetes cure in the recipient and the absence of diabetes in the donor [38]. Furthermore, normal rats 93that underwent a 60% pancreatectomy remained normoglycaemic due to partial beta-cell regeneration and hyperfunction of remaining beta cells [39]. However, when these rats were challenged by the addition of 10% sugar to their water supply, impairment of the residual beta-cell mass and subsequent hyperglycaemia ensued. Therefore, in isolation, reduced beta-cell mass was insufficient to cause diabetes but rather required the presence of environmental factors to unmask the effect.
There are well-described functional defects in the beta cells of subjects with diabetes. As mentioned earlier, the loss of first phase insulin secretion is a particularly early defect [12]. Another is the loss of oscillations in insulin secretion, the importance of which relates to the regulation of hepatic glucose production under physiological conditions [40]. This may occur early in the disease, being found in non-diabetic relatives of people with type 2 diabetes [41]. Thirdly, inefficient conversion of the precursor peptide, pro-insulin, into insulin results in a four to fivefold increase in pro-insulin levels in the beta cells of patients with type 2 diabetes [42].
Defects within the incretin system may also result in early beta-cell dysfunction [14]. The incretins are a pair of gut hormones, namely glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide, which are released into the bloodstream after meal ingestion and stimulate insulin secretion in a glucose-dependent manner. The incretins increase the expression of genes important for insulin secretion, such as the GLUT2 and glucokinase genes, and beta-cell proliferation, such as PDX-1. Subjects with type 2 diabetes may display down-regulation of incretin receptors on the surface of beta cells [43], impaired incretin-induced insulin secretion [44] and increased degradation of incretin hormones [45]. Non-diabetic relatives of those with diabetes [46] and people with impaired glucose tolerance [47] may also display defects in the incretin system.
A whole range of gene expression defects are also associated with beta-cell dysfunction. My research group have previously demonstrated that proteins vital to beta-cell function and insulin secretion, known as the transcription factors HIF-1a [48] and ARNT [49], have significantly less expression in the pancreatic islets of diabetic versus non-diabetic subjects. Knockout mice lacking the genes for these proteins in insulin expressing cells develop glucose intolerance and impaired insulin secretion [44, 45]. In fact, the advent of genome-wide association studies (GWAS) in 2007 has led to an explosion of data on the possible genetic background of type 2 diabetes [20]. GWAS uses thousands of small nucleotide sequences throughout the genome to search for patterns linked to disease, using the human genome sequencing project as reference. The transcription factor 7-like 2 (TCF7L2) is to be most strongly linked to type 2 diabetes with important effects on beta-cell proliferation and function [50], incretin-related insulin secretion [39] and insulin secretion [51]. Polymorphisms in TCF7L2 are thought to account for ~20% of the population attributable risk of diabetes [52]. The important point here is that the genetic determinants of beta-cell function and mass appear closely linked.
Some investigators now refer to ‘reduced functional beta-cell mass’ rather than debating the predominance of reduced mass or defective function in diabetes causation [53]. The 94‘plasticity’ of the functional beta-cell mass, that is, the concept that beta cells can undergo adaptive changes in both mass and function when challenged by insulin resistance has also received some attention.
Mechanisms of beta-cell failure
If defects in the functional beta-cell mass play such a critical role in diabetes pathogenesis, what are some of the mechanisms responsible for these defects? These have been summarised in Figure 3.
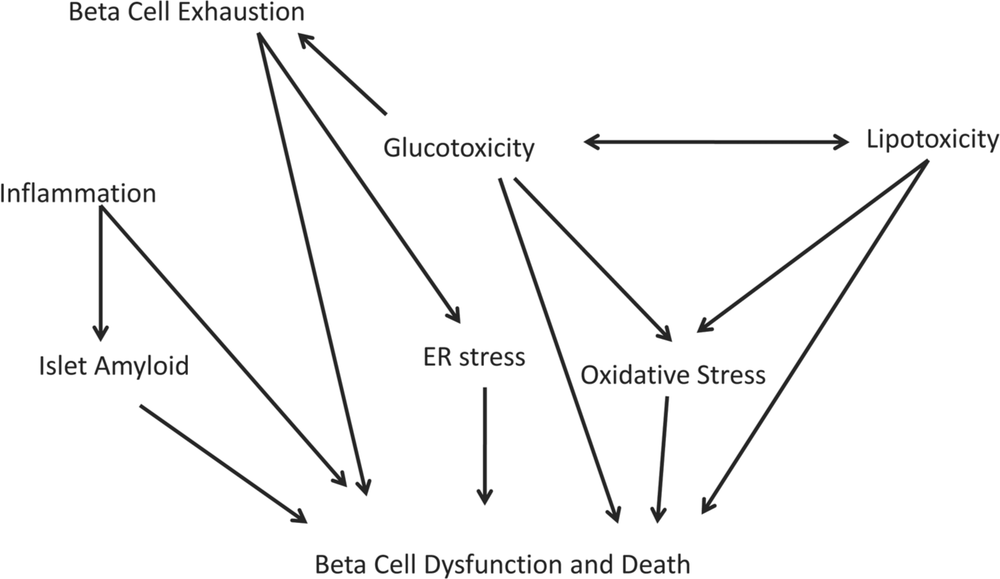
Figure 3. Proposed mechanisms of pancreatic beta (beta) cell dysfunction and death in type 2 diabetes. This flow-chart depicts the complex and multifactorial nature of cell dysfunction in this condition. For a description of the concepts and terms see the text.
Pancreatic islet inflammation
There is mounting evidence that chronic low-grade inflammation of the pancreatic islet can cause insulin secretory dysfunction and beta-cell apoptosis [54]. Most striking is the macrophage infiltration seen in the islets of mice and humans with type 2 diabetes prior to the onset of beta-cell death [55]. It is also known that islets and ductal cells respond to metabolic stress, such as hyperglycaemia [56] and exposure to fatty acids, by expressing cytokines particularly IL-1beta. While this may initially be protective, chronic expression of this cytokine may result in the production of reactive oxygen species, inflammation and eventual beta-cell death [49]. Furthermore, the use of anti-inflammatory therapies, such as an IL-1 receptor antagonists [57] or non-steroidal anti-inflammatory drugs [58] results in small improvements in beta-cell function independent of an effect on insulin resistance. 95Inflammatory markers, such as the serum leukocyte count and c-reactive protein, are also elevated in patients with type 2 diabetes and in those at risk of diabetes [59].
There is emerging evidence linking obesity and beta-cell failure via an inflammatory pathway. Leptin, a hormone secreted by fat cells to control appetite, is often found in high levels in type 2 diabetes [60] and at such levels, has negative effects on insulin secretion [61] and may induce beta-cell apoptosis [62]. Conversely, adiponectin which has anti-inflammatory and insulin sensitising effects, appears to be down-regulated in diabetes [63]. In addition, fat-derived cytokines, fatty acids and lipoproteins may all have adverse effects on beta-cell function, suggesting a common inflammatory link between the various components of the metabolic syndrome.
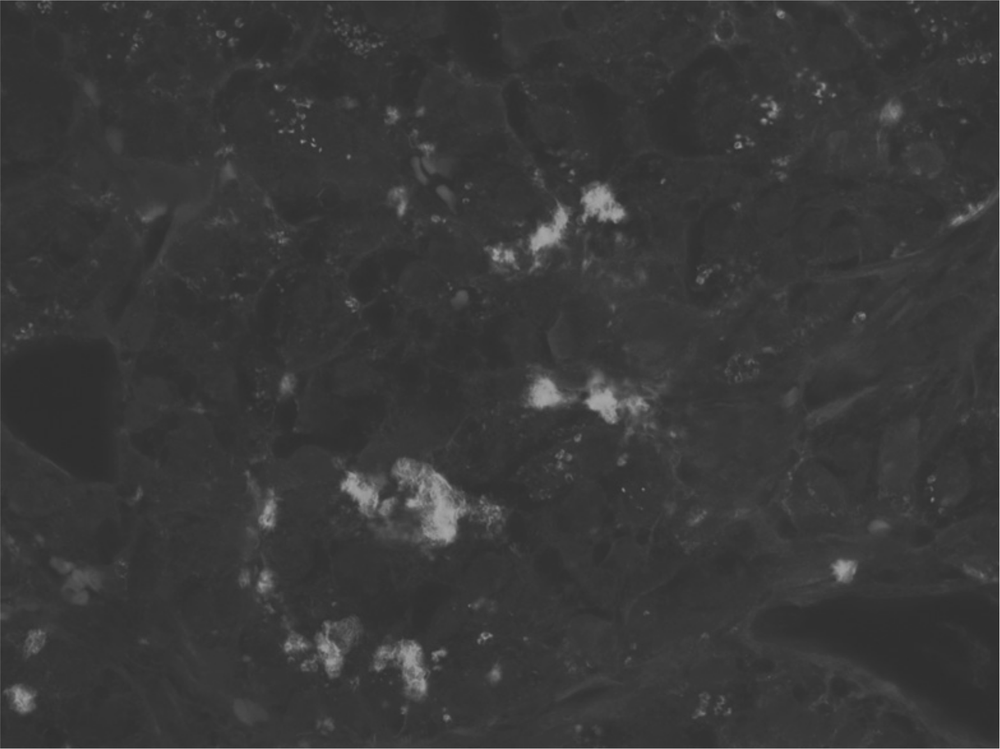
Figure 4. Islet amyloid deposition in a human with type 2 diabetes In the islet of this patient with diabetes, amyloid infiltration is seen as bright plaques in this grey scale image, thioflavin S stain.
Islet amyloid
Although islet amyloidosis has long been associated with beta-cell loss in type 2 diabetes [64], its precise role is uncertain. At postmortem, more than 90% of type 2 diabetic subjects have amyloid deposits in at least one islet compared to 12% of age-matched non-diabetic 96patients [65]. However, the number of islets affected is highly variable [66] and there is no relationship between duration of diabetes and the presence or severity of islet amyloid [67].
Islet amyloid fibrils are composed of amylin, a normally occurring peptide of the beta cell co-stored with insulin, which has folded abnormally and is initially deposited around islet capillaries but later within the islet space [30]. The increased production of pro-amylin accompanies the higher pro-insulin levels characteristic of type 2 diabetes and may be the first step to developing islet amyloid [68].
Although synthetic amyloid fibrils induce early beta-cell death in vitro [69], it is unclear whether amyloidosis is the direct cause of beta-cell failure in vivo or rather, the end-result of several distinct processes including inflammation, oxidative stress and mitochondrial damage. It is also currently debated whether the amyloid plaques per se are cytotoxic or rather small intracellular molecules that are precursors in the development of such plaques [70].
Endoplasmic reticulum stress
The endoplasmic reticulum (ER), a highly developed organelle that resides within the cell, is responsible for protein handling and packaging. Due to the high secretory demand placed on beta cells in type 2 diabetes, the endoplasmic reticulum may become overwhelmed by the production of high level of proteins [71]. A process called ER stress ensues whereby intracellular signals intending to protect the beta cells from the effects of inflammation [72] may eventually lead to the impairment of beta-cell function, the morphological appearance of ER swelling as proteins accumulate and subsequent beta-cell death.
ER stress may be mediated via genetic and environmental factors. Mice with mutations in proinsulin develop diabetes with morphological evidence of ER stress [20]. Exposure of islets to high glucose levels, cytokines and fatty acids may also lead to signs of ER stress [73].
Glucose toxicity
Glucose is the key physiological regulator of insulin secretion. Chronic hyperglycaemia is associated with impaired insulin secretion [74] and restoring normoglycaemia in subjects with diabetes has a clearly positive effect on beta-cell function [75].
There are several ways by which the exposure of beta cells to high glucose levels has toxic effects (glucotoxicity). The term ‘beta-cell dedifferentiation’ refers to an altered pattern of gene expression that results in substantial functional defects related to chronic hyperglycaemia [76]. Changes in beta-cell signalling and architecture are also possible effects of high glucose exposure [77]. The overproduction of reactive oxygen species, known as oxidative stress, is related to mitochondrial metabolism of high glucose levels and may result in beta-cell dysfunction or death [20].
Support for the oxidative stress hypothesis has come from a number of animal and human studies. The beta-cell over-expression of glutathione peroxidase, the enzyme that protects 97cells from oxidative damage, improved beta-cell mass and function in diabetic mice [78]. Higher concentrations of oxidative stress markers were found in islets from human subjects with type 2 diabetes and marked improvement in insulin secretion followed 24 hours of exposure to glutathione [79].
The failure of the beta cell’s adaptive response to high glucose levels may precede dysfunction. The Fas-FLIP pathway involves a series of factors which are upregulated in response to high glucose levels and which result in greater beta-cell differentiation, proliferation and function [80]. However, in chronic hyperglycaemia, this adaptive pathway may eventually fail resulting in deterioration in beta-cell function and reduced survival [49].
Lipotoxicity
Whilst effective insulin secretion requires the presence of fatty acids [81], long-term exposure to excessive levels of this nutrient may also result in beta-cell toxicity [30]. The precise mechanism of so-called lipotoxicity is unclear but it may be due to genetic alterations in the expression of insulin and enzymes controlling insulin secretion [82], metabolic disturbances and ultimately, beta-cell death.
Studies in the Zucker diabetic fatty rat, considered a good model for obesity-related type 2 diabetes, have shown that the development of hyperlipidaemia parallels inadequate insulin secretion and the onset of diabetes [83]. However the lipid levels in this animal model are often extreme, so whether this reflects the human condition is debatable. Furthermore, fewer than 12% of obese hyperlipidaemic human subjects become diabetic [84], suggesting that lipotoxicity may be just one component in the development of beta-cell failure in type 2 diabetes.
Beta-cell exhaustion
It is speculated that after a prolonged period of insulin hypersecretion, depletion of readily available insulin granules may occur, thereby reducing the insulin secretory response [20]. Although we previously discussed the limitations of beta-cell exhaustion as the sole pathogenic mechanism, it may play a role in the wider, multifactorial setting of diabetes pathogenesis.
The strongest support for the exhaustion concept comes from studies examining the benefits of ‘beta-cell rest’ strategies. When inhibitors of insulin secretion, such as diazoxide or somatostatin, are given to animals or humans with diabetes, paradoxical increases in beta-cell function are observed following a period of ‘rest’ [85, 86]. Furthermore, human subjects with poorly controlled diabetes demonstrate a dramatic improvement in beta-cell function following a brief period of intensive glycaemic control [67]. A multicentre, randomised study of over 350 newly diagnosed patients confirmed that early intensive insulin therapy may facilitate recovery and maintenance of beta-cell function and protracted glycaemic remission better than oral hypoglycaemic agents for at least one year after diagnosis [87].98
This leads to the discussion of the therapeutic implications of beta-cell failure and the possibility of reversing this complex process. Although a highly controversial and evolving area of research, the use of certain sulfonylureas such as glibenclamide has been linked to beta-cell death [88], while there is mounting evidence that incretin-based therapy and insulin replacement increase beta-cell mass in mice and have a positive effect on function [89]. However, a discussion on the nuances of beta-cell preservation in type 2 diabetes is beyond the scope of this chapter. For such a discussion, we refer the reader to a recent review on this topic [81].
Conclusion
Pancreatic beta-cell dysfunction plays a vital and complex role in the pathogenesis of type 2 diabetes. Ultimately, it is the failure of beta cells to respond to the insulin requirements posed by the prevailing level of insulin resistance that leads to the development of type 2 diabetes. We have discussed the evidence in support of various genetic and environmental factors that conspire together in this state of beta-cell susceptibility and ‘decompensation’. The precise role of each and the search for a hypothesis which may unify these disparate concepts into a single paradigm is highly contentious and under active investigation. However, there is no doubt that beta-cell demise occurs in stages with early defects in beta-cell dysfunction, such as loss of first phase insulin secretion resulting in post-prandial hyperglycaemia following which progressive rises in glucose levels, fatty acids and insulin resistance place further stress on the beta cell, leading to eventual decline in its secretory capacity. Subtle components of the beta-cell stress mechanism, initially adaptive, such as changes to the endoplasmic reticulum and mitochondrium also eventually exacerbate the situation. Recent evidence suggests that early intervention to improve glycaemic control may improve beta-cell function and for at least a limited time period, may retard progression to beta-cell failure. Further research into the complex mechanisms of beta-cell failure may therefore offer hope in our collective fight against the global epidemic of type 2 diabetes.
References
1. Glatthaar C, Welborn TA, Stenhouse NS & Garcia-Webb P (1985). Diabetes and impaired glucose tolerance: a prevalence estimate based on the Busselton 1981 survey. The Medical Journal of Australia, 143(10): 436–40.
2. Dunstan DW, Zimmet PZ, Welborn TA, DeCourten MP, Cameron AJ & Sicree RA, et al. (2002). The rising prevalence of diabetes and impaired glucose tolerance: the Australian diabetes, obesity and lifestyle study (AusDiab). Diabetes Care, 25(5): 829–34.
3. Wild S, Roglic G, Green A, Sicree R & King H (2004). Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care, 27(5): 1047–53.
4. Yang W, Lu J, Weng J, Jia W, Ji L, Xiao J & Shan Z, et al. (2010). Prevalence of diabetes among men and women in China. New English Journal of Medicine, 362: 1090–101.99
5. Diabetes Australia (2010). Diabetes in Australia. [Online]. Available: www.diabetesaustralia.com.au/Understanding-Diabetes/Diabetes-in-Australia [Accessed 26 September 2011].
6. Sakula A (1988). Paul Langerhans: a centenary tribute. Journal of the Royal Society of Medicine, 81(7): 414–15.
7. Bonner-Weir S (1991). Anatomy of islet of Langerhans. In Samols E (Ed). The endocrine pancreas (pp15–27). New York: Raven.
8. Rahier J, Guiot Y, Goebbels RM, Sempoux C & Henquin JC (2008). Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes, Obesity and Metabolism, 10(Suppl. 4): 32–42.
9. Klöppel G, Löhr M, Habich K, Oberholzer M & Heitz PU (1985). Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Survey and Synthesis of Pathology Research, 4(2): 110–25.
10. Lifson N, Lassa CV & Dixit PK (1985). Relation between blood flow and morphology in islet organ of rat pancreas. American Journal of Physiology, 249(1 Pt 1): E43–48.
11. Kaido T, Yebra M, Cirulli V, Rhodes C, Diaferia G & Montgomery AM (2006). Impact of defined matrix interactions on insulin production by cultured human beta-cells: effect on insulin content, secretion and gene transcription. Diabetes, 55(10): 2723–29.
12. Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B & Harlan DM, et al. (2005). Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. Journal of Histochemistry and Cytochemistry, 53(2): 1087–97
13. Straub SG, James RF, Dunne MJ & Sharp GW (1998). Glucose activates both K(ATP) channel-dependent and K(ATP) channel-independent signaling pathways in human islets. Diabetes, 47(5): 758–63.
14. Perley MJ & Kipnis DM (1967). Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic subjects. Journal of Clinical Investigation, 46(12): 1954–62.
15. Klee P, Bavamian S, Charollais A, Caille D, Cancela J, Peyrou M, et al. (2008). Gap junctions and insulin secretion. In Seino S & Bell GI (Eds). Pancreatic beta cell in health and disease (pp111–32). Japan: Springer.
16. Ahren B (2006). Incretins and islet function. Current Opinion in Endocrinology & Diabetes, 13(2): 154–61.
17. Ahren B (2009). Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nature Reviews Drug Discovery, 8: 369–85.
18. Nolan CJ & Prentki M (2008). The islet beta-cell: fuel responsive and vulnerable. Trends in Endocrinology and Metabolism, 19(8): 285–91.
19. Lau SL, Clifton-Bligh R, Eismann J & Gunton JE (2008). A functional and regulated vitamin D system exists within Min-6 cells, an immortalized mouse-derived beta-cell line. Australian Diabetes Society Annual Scientific Meeting.
20. Baetens D, Malaisse-Lagae F, Perrelet A & Orci L (1979). Endocrine pancreas: three-dimensional reconstruction shows two types of islets of Langerhans. Science, 206(4424): 1323–25.100
21. Haataja L, Gurlo T, Huang CJ & Butler PC (2008). Islet amyloid in type 2 diabetes and the toxic oligomer hypothesis. Endocrinology Review, 29(3): 303–16.
22. Leahy JL & Pratley RE (2011). What is type 2 diabetes mellitus? Crucial role of maladaptive changes in beta cell and adipocyte biology In Robertson RP & Powers AC (Eds). Translational endocrinology and metabolism: Type 2 Diabetes (pp9–43). Maryland: The Endocrine Society.
23. Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS & Kahn CR (1992). Role of glucose and insulin resistance in the development of type 2 diabetes mellitus: results of a 25-year follow-up study. The Lancet, 340(8825): 925–29.
24. Lillioja S, Mott DM, Howard BV, Bennett PH, Yki-Jarvinen H, Freymond D, et al. (1988). Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. The New English Journal of Medicine, 318(19): 1217–25.
25. Weir GC & Bonner-Weir S (2004). Five stages of evolving beta cell dysfunction during progression to diabetes. Diabetes, 53(S3): 16–21.
26. Hansen BC & Bodkin NL (1990). Beta-cell hyperresponsiveness: earliest event in the development of diabetes in monkeys. The American Journal of Physiology, 259(3): R612–17.
27. Krook, A & O’Rahilly S (1996). Mutant insulin receptors in syndromes of insulin resistance. Baillieres Clinical Endocrinology and Metabolism, 10(1): 97–122.
28. Weyer C, Bogardus C, Mott DM & Pratley RE (1999). The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. Journal of Clinical Investigation, 104(6): 787–94.
29. Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW, et al. (1993). Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects: evidence for a hyperbolic function. Diabetes, 42(11): 1663–72.
30. Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, Schubert M, Fisher TL, et al. (2003). Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. Journal of Clinical Investigation, 112(10): 1521–32.
31. George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S & Wilson JC (2004). A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science, 304(5675): 1325–28.
32. Shoelson SE, Lee J & Yuan M (2003). Inflammation and the IKK beta/I kappa B/NF kappa B axis in obesity- and diet-induced insulin resistance. International Journal of Obesity and Related Metabolic Disorders, 27(Suppl. 3): S49–52.
33. Donath MY, Storling J, Maedler K & Mandrup-Poulsen T (2003). Inflammatory mediators and islet beta cell failure: a link between type 1 and type 2 diabetes. Journal of Molecular Medicine, 81(8): 455–70.
34. Clark A (2008). Pancreatic islet pathology in type 2 diabetes. In Seino S & Bell GI (Eds). Pancreatic beta cell in health and disease (pp381–98). Japan: Springer.
35. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA & Butler PC (2003). Beta cell deficit and increased beta cell apoptosis in humans with type 2 diabetes. Diabetes, 52(1): 102–10.101
36. Powers AC, Stein RW (2011). In Robertson RP & Powers AC (Eds). Translational endocrinology and metabolism: type 2 diabetes (pp95–116). Maryland: The Endocrine Society.
37. MacLean N & Ogilvie RF (1955). Quantitative estimation of pancreatic islet tissue in diabetic subjects. Diabetes, 4(5): 367–76.
38. Matsumoto S, Okitsu T, Iwanaga Y, Noguchi H, Nagata H, Yonekawa Y, et al. (2005). Insulin independence of unstable diabetic patient after single living donor islet transplantation. Transplant Proceeding, 37(8): 3427–29.
39. Leahy JL, Bonner-Weir S & Weir GC (1988). Minimal chronic hyperglycaemia is a critical determinant of impaired insulin secretion after an incomplete pancreatectomy. Journal of Clinical Investigation, 81(5): 1407–14.
40. Porkson N (2002). The in vivo regulation of pulsatile insulin secretion. Diabetologia, 45(1): 3–20.
41. O’Rahilly S, Turner RC & Matthews DR (1988). Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. The New England Journal of Medicine, 318(19): 1225–30.
42. Kahn SE & Halban PA (1997). Release of incompletely processed pro-insulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes, 46(1): 1725–32.
43. Shu L, Matveyenko AV, Kerr-Conte J, Cho JH, McIntosh CH & Maedler K (2009). Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Human Molecular Genetics, 18(13): 2388–99.
44. Meier JJ, Gallwitz B, Kask B, Deacon CF, Holst JJ, Schmidt WE, et al. (2004). Stimulation of insulin secretion by intravenous bolus injection and continuous infusion of gastric inhibitory polypeptide in patients with type 2 diabetes and healthy control subjects. Diabetes, 53(Suppl. 3): S220–24.
45. Mannucci E, Pala L, Ciani S, Bardini G, Pezzatini A, Sposato I, et al. (2005). Hyperglycaemia increases dipeptidyl peptidase IV activity in diabetes mellitus. Diabetologia, 48(6): 1168–72.
46. Meier JJ, Hucking K, Holst JJ, Deacon CF, Schmiegel WH & Nauck MA (2001). Reduced insulinotropic effect of gastric inhibitory polypeptide in first-degree relatives of patients with type 2 diabetes. Diabetes, 50(11): 2497–04.
47. Faerch K, Vaag A, Holst JJ, Glumer C, Pederson O & Borch-Johnsen K (2008). Impaired fasting glycaemia vs impaired glucose tolerance: similar impairment of pancreatic alpha and beta-cell function but differential roles of incretin hormones and insulin action. Diabetologia, 51(5): 853–61.
48. Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ et al. (2010). Hypoxia-inducible factor-1alpha regulates beta-cell function in mouse and human islets. Journal of Clinical Investigation, 120(6): 2171–83.
49. Gunton JE, Kulkarni RN, Yim S, Okada T, Hawthorne WJ, Tseng Y, et al. (2005). Loss of ARNT/HIF1β mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes cell. Cell Metabolism, 122: 337–49.
50. Liu Z & Habener JF (2010). Wnt signaling in pancreatic islets. Advances in Experimental Medicine and Biology, 654: 391–419.102
51. da Silva Xavier G, Loder MK, McDonald A, Tarasov AI, Carzaniga R, Kronenberger K, et al. (2009). TCF7L2 regulates late events in insulin secretion from pancreatic islet beta cells. Diabetes, 58(4): 894–905.
52. Grant SFA, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, et al. (2006). Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nature Genetics, 38: 320–23.
53. Karaca M, Magnan C & Kargar C (2009). Functional pancreatic beta-cell mass: involvement in type 2 diabetes and therapeutic intervention. Diabetes and Metabolism, 35: 77–84.
54. Donath MY & Ehses JA (2008). Mechanisms of beta cell death in diabetes. In Seino S & Bell GI (Eds). Pancreatic beta cell in health and disease (pp75–89). Japan: Springer.
55. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. (2007). Increased number of islet associated macrophages in type 2 diabetes. Diabetes, 56: 2356–70.
56. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. (2002). Glucose-induced beta-cell production of interleukin-1beta contributes to glucotoxicity in human pancreatic islets. Journal of Clinical Investigation, 110: 851–60.
57. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. (2007). Interleukin-1-receptor antagonist in type 2 diabetes mellitus. The New England Journal of Medicine, 356: 1517–26.
58. Zeender E, Maedler K, Bosco D, Berney T, Donath MY & Halban PA (2004). Pioglitazone and sodium salicylate protect human beta cells against apoptosis and impaired function induced by glucose and interleukin-1beta. Journal of Clinical Endocrinology and Metabolism, 89: 5059–66.
59. Pham MN, Hawa MI, Pfleger C, Roden M, Schernthaner G, Pozzilli P, et al. (2011). Pro- and anti-inflammatory cytokines in latent autoimmune diabetes in adults, type 1 and type 2 diabetes patients: action LADA 4. Diabetologia, 54(7): 1630–38.
60. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. (2001). Hypoadiponectinaemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinaemia. Journal of Clinical Endocrinology and Metabolism, 86: 1930–35.
61. Kieffer TJ & Habener JF (2000). The adipoinsular axis: effects of leptin on pancreatic beta cells. American Journal of Physiology: Endocrinology and Metabolism, 278: E1–14.
62. Maedler K, Sergeev P, Ehses JA, Mathe Z, Bosco D, Berney T, et al. (2004). Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proceedings of the National Academy of Sciences USA, 101: 8138–43.
63. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE et al. (2001). Hypoadiponectinaemia in obesity and type 2 diabetes: a close association with insulin resistance and hyperinsulinaemia. Journal of Clinical Endocrinology and Metabolism, 86: 1930–35.
64. Wright A (1927). Hyaline degeneration of the islets of Langerhans in non-diabetics. American Journal of Pathology, 3(5): 461–82.
65. Rocken C, Linke RP & Saeger W (1992). Immunohistology of islet amyloid polypeptide in diabetes mellitus: semi-quantitative studies in a post-mortem series. Virchows Archive A: Pathological Anatomy and Histopathology, 421: 339–44.103
66. Clark A, Holman R, Matthews D, Hockaday T & Turner R (1984). Non-uniform distribution of islet amyloid in the pancreas of ‘maturity-onset’ diabetic patients. Diabetologia, 27: 527–28.
67. Westermark P (1984). Amyloid and polypeptide hormones: what is their interrelationship? Amyloid: International Journal of Experimental and Clinical Investigation, 1: 47–57.
68. Paulsson JF & Westermark GT (2005). Aberrant processing of human proislet amyloid polypeptide results in increased amyloid formation. Diabetes, 54: 2117–25.
69. Marzban L, Rhodes CJ, Steiner DF, Haataja L, Halban PA & Verchere CB (2006). Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes, 55: 2192–201.
70. Zraika S, Hull RL, Verchere CB, Clark A, Potter KJ, Fraser PE, et al. (2010). Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence? Diabetologia, 53: 1046–56.
71. Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. (2007). Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia, 50: 752–63.
72. Weber SM, Chambers KT, Bensch KG, Scarim AL & Corbett JA (2004). PPARgamma ligands induce ER stress in pancreatic beta cells: ER stress activation results in attenuation of cytokine signaling. American Journal of Physiology: Endocrinology and Metabolism, 287: E1171–77.
73. Marchetti P, Bugliani M Lupi R, Marselli l, Masini M, Boggi U, et al. (2007). The endoplasmic reticulum in pancreatic beta cell of type 2 diabetes patients. Diabetologia, 50: 2486–94.
74. Unger RH & Grundy S (1985). Hyperglycaemia as an inducer as well as a consequence of impaired islet cell function and insulin resistance: implications for the management of diabetes. Diabetologia, 28: 119–21.
75. Turner RC, McCarthy ST, Holman RR & Harris E (1976). Beta-cell function improved by supplementing basal insulin secretion in mild diabetes. British Medical Journal, 1: 1252–54.
76. Leahy JL, Cooper HE, Deal DA & Weir GC (1986). Chronic hyperglycaemia is associated with impaired glucose influence on insulin secretion: a study in normal rats using chronic in vivo glucose infusion. Journal of Clinical Investigation, 77: 908–15.
77. Leahy JL (2004). Detrimental effects of chronic hyperglycemia on the pancreatic β-cell. In LeRoith D, Olefsky JM & Taylor S (Eds). Diabetes mellitus: a fundamental and clinical text (pp115–27). Philadelphia: Lippincott.
78. Harmon JS, Bogdani M, Parazzoli SD, Mark SS, Oseid EA, Berghmans M, et al. (2009). Beta-cell specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology, 150: 4855–62.
79. Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, et al. (2005). Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes, 54: 727–35.
80. Donath MY, Ehses JA, Maedler K, Schumann DM, Ellingsgaard H, Eppler E, et al. (2005). Mechanisms of beta cell death in type 2 diabetes. Diabetes, 54(Suppl. 2): S108–13.104
81. Dobbins RL, Chester MW, Stevenson BE, Daniels MB, Stein DT & McGarry JDI (1998). A fatty acid – dependent step is critically important for both glucose- and non-glucose-stimulated insulin secretion. Journal of Clinical Investigation, 101: 2370–76.
82. Kebede M, Favaloro J, Gunton JE, Laybutt DR, Shaw M, Wong N, et al. (2008). Fructose-1,6-bisphosphatase overexpression in pancreatic beta-cells results in reduced insulin secretion: a new mechanism for fat-induced impairment of beta-cell function. Diabetes, 57: 1887–95.
83. Unger RH & Zhou YT (2001). Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes, 50(Suppl. 1): S118–21.
84. Agren G, Narbro K, Naslund, Sjostrom L & Peltonen M (2002). Long-term effects of weight loss on pharmaceutical costs in obese subjects: a report from the SOS intervention study. International Journal of Obesity Related Metabolism Disorders, 26: 184–92.
85. Leahy JL, Bumbalo LM & Chen C (1994). Diazoxide causes recovery of beta-cell glucose responsiveness in 90% pancreatectomised diabetic rats. Diabetes, 44: 173–79.
86. Greenwood RH, Mahler RF & Hales CN (1976). Improvement in insulin secretion in diabetes after diazoxide. The Lancet, 1: 444–47.
87. Weng J, Li Y, Xu W, Shi L, Zhang Q, Zhu D, et al. (2008). Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. The Lancet, 371: 1753–60.
88. Efanova IB, Zaitsev SV, Zhivotovsky B, Kohler M, Efendic S, Orrenius S, et al. (1998). Glucose and tolbutamide induce apoptosis in pancreatic beta cells: a process dependent on intracellular xcalcium concentration. The Journal of Biological Chemistry, 273: 33501–07.
89. Leahy JL, Hirsch IB, Peterson KA & Schneider D (2010). Targeting beta-cell function early in the course of therapy for type 2 diabetes mellitus. Journal of Clinical Endocrinology and Metabolism, 95: 4206–16.